Abstract
Saposhnikovia divaricata, a well-known Chinese medicinal herb, is the sole species under the genus Saposhnikovia of the Apiaceae subfamily Apioideae Drude. However, information regarding its genetic diversity and evolution is still limited. In this study, the first complete chloroplast genome (cpDNA) of wild S. divaricata was generated using de novo sequencing technology. Similar to the characteristics of Ledebouriella seseloides, the 147,834 bp-long S. divaricata cpDNA contained a large single copy, a small single copy, and two inverted repeat regions. A total of 85 protein-coding, 8 ribosomal RNA, and 36 transfer RNA genes were identified. Compared with five other species, the non-coding regions in the S. divaricata cpDNA exhibited greater variation than the coding regions. Several repeat sequences were also discovered, namely, 33 forward, 14 reverse, 3 complement, and 49 microsatellite repeats. Furthermore, phylogenetic analysis using 47 cpDNA sequences of Apioideae members revealed that L. seseloides and S. divaricata clustered together with a 100% bootstrap value, thereby supporting the validity of renaming L. seseloides to S. divaricata at the genomic level. Notably, S. divaricata was most closely related to Libanotis buchtormensis, which contradicts previous reports. Therefore, these findings provide a valuable foundation for future studies on the genetic diversity and evolution of S. divaricata.
1. Introduction
Saposhnikovia divaricata (Turcz.) Schischk., the sole species of the genus Saposhnikovia Schischk, under the Apiaceae subfamily Apioideae Drude, is widely distributed in the Northern regions of China. It is one of the most important and well-known traditional Chinese medicinal plants listed in the Chinese Pharmacopoeia, as well as in several pharmaceutical records, such as the Thousand Golden Prescriptions (Qian Jin Fang) and Shen Nong’s Materia Medica (Shen Nong Ben Cao Jing). The dried roots are called Fang-Feng in China, Bang-Poong in Korea, and Bofu in Japan, and have been extensively used for treating arthralgia, headaches, rheumatism, stroke, fever, and allergic rhinitis [1]. Recently, studies investigating the chemical constituents of S. divaricata revealed that the main active components were chromones, coumarins, and volatile oils [2,3,4], which exhibited anti-proliferative and anti-oxidant, anti-bacterial and anti-tumor, anti-convulsant, anti-coagulant, anti-inflammatory, and anti-pyretic properties [4,5,6,7,8]. However, little information is known regarding genetic diversity and evolution.
The chloroplast is a photosynthetic organelle in algae and plants that provides the energy essential for growth and reproduction by promoting the biosynthesis and metabolism of starch and fatty acids [9]. Recent studies have shown that this double membrane plant organelle originated from the endosymbiosis of cyanobacteria [10]. Chloroplast genomes (cpDNAs) are maternally inherited in most plants, and the majority of angiosperm cpDNAs are characterized by small molecules, high copy number genes, and highly conserved sequences [11,12]. Typically, cpDNAs are closed circular double-stranded DNA with a classic quadripartite structure composed of two inverted repeat regions (IRa and IRb), a small single copy (SSC) region, and a large single copy (LSC) region [13]. The cpDNA composition and sequence in angiosperms consist of highly conserved protein-coding genes (PCGs), transfer RNA (tRNA) genes, and ribosomal RNA (rRNA) genes [14]. However, the size, structure, and IR contraction and expansion of angiosperm cpDNAs have undergone several alterations caused by the adaptation to changing environments, and pseudocolonization even occurred in some genera [15]. Therefore, cpDNAs can be used to analyze the genetic structure and molecular characteristics among closely related plant species. The sequenced cpDNAs are generally available to download in public databases, such as the National Center for Biotechnology Information (NCBI; https://www.ncbi.nlm.nih.gov/; accessed on 14 September 2020).
With the rapid development of next-generation sequencing technology, the cpDNAs of numerous species have been fully sequenced and functionally characterized. However, although the cpDNA sequences of the cultivated S. divaricata in China [16,17] and its synonymous Ledebouriella seseloides from South Korea [18] have been reported, little is known about the cpDNA information of wild S. divaricata, especially its genetic diversity and evolutionary relationship with L. seseloides and other related species. L. seseloides has already been renamed S. divaricata [19], however, it is still used by some researchers [18]. Furthermore, the cpDNA sequences of L. seseloides and S. divaricata were separately published and have not yet been analyzed in one study. The genomic analysis of available data for these species will further support the validity of this renaming at the molecular level. To investigate the genetic characteristics and phylogeny of wild S. divaricata and to discover a molecular basis for the renaming of L. seseloides, we collected wild S. divaricata samples (33°72′ N, 112°02′ E) for high-throughput cpDNA sequencing and conducted an in-depth analysis via comparison with L. seseloides and other related species. Specifically, we aimed to determine the genomic features of the wild S. divaricata cpDNA to extensively compare its cpDNA with L. seseloides and other Apioideae subfamily members and to identify the repeats and simple sequence repeats (SSRs), thereby discovering the unique characteristics of the wild S. divaricata cpDNA. The comparison between the cpDNA sequences of wild S. divaricata and 46 other taxa under the subfamily Apioideae revealed their phylogenetic relationships. The repeats identified in this study may be useful for developing SSR markers to analyze the genetic diversity of Apioideae subfamily members and are candidates for DNA barcoding studies. Our findings may provide a foundation for future genomic research on the genetic diversity and evolution of S. divaricata and other related species.
2. Results and Discussion
2.1. Genomic Features of the Wild S. divaricata CpDNA
The 147,834 bp-long wild S. divaricata cpDNA was composed of a 93,202 bp-long LSC, a 17,324 bp-long SSC, and a pair of 18,654 bp-long IR (Figure 1) regions. The IRa and IRb regions contained genes of the same type but were arranged in reverse. The length of the SSC region in wild S. divaricata cpDNA was similar to those of other herbs (17,000–19,000 bp), but the lengths of the IR regions were significantly shorter than those of other herbs [20,21]. The GC content of the wild S. divaricata cpDNA was 37.5%, which is consistent with previous reports [16,17]. Determining the GC content in the four regions is necessary for exploring species evolution and genetic relationships and is considered an important parameter for evaluating the codon preference and evolutionary trend in plants. Similar to other closely related species, the GC content in the IR regions of wild S. divaricata cpDNA was 44.6%, which is higher than those in the LSC (35.9%) and SSC (36.0%) regions (Table S1, Figure 2). In addition, we re-annotated, analyzed, and compared all the reported cpDNA sequences of S. divaricata and its synonymous species, L. seseloides. The results showed that the main difference between the two was the cpDNA size, although the total number of genes and unigenes was the same (Table S2).
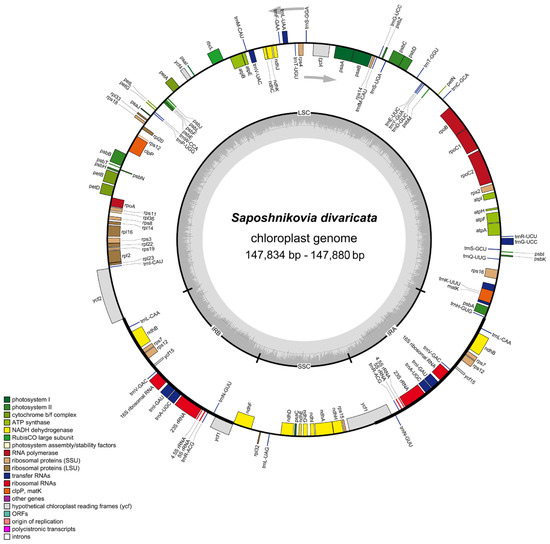
Figure 1.
Chloroplast genome map showing all reported genes of Saposhnikovia divaricata. Genes drawn inside the circle are transcribed clockwise, and those outside are counterclockwise. Genes belonging to different functional groups are color-coded. The darker gray in the inner circle corresponds to GC content. Small single-copy (SSC) region, large single-copy (LSC) region, and inverted repeats (IRa and IRb) are displayed.
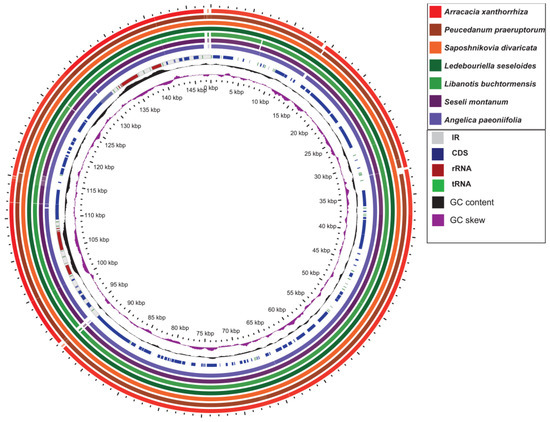
Figure 2.
Comparison of GC content of the wild S. divaricata cpDNA using GView program.
The comprehensive and in-depth analysis of wild S. divaricata cpDNA revealed 129 functional genes, including 8 rRNA genes, 36 tRNA genes, and 85 PCGs (Table 1). Except for the double-copy gene rps12 located in the LSC and IR regions, all genes, including eight tRNA genes (trnA-UGC, trnG-UCC, trnI-GAU, trnL-CAA, trnL-UAA, trnN-GUU, trnR-ACG, and trnV-GAC), five PCGs (rps7, rps12, ndhB, ycf1, and ycf15), and four rRNA genes (rrn4.5, rrn5, rrn16, and rrn23) were duplicated in the IR regions. Additionally, the LSC region contained 24 tRNA genes and 66 PCGs. By contrast, the SSC region only possessed one tRNA and twelve PCGs. Notably, five types of ycf (ycf1, ycf2, ycf3, ycf4, and ycf15) were detected in this genome. Moreover, two genes (clpP and ycf3) contained two introns (rps12 was special with two copies, and its first exon was shared in the LSC region, and exons 2 and 3 were in the IR region), whereas six tRNAs (trnK-UUU, trnI-GAU, trnA-UGC, trnG-UCC, trnV-UAC, and trnL-UAA) and nine PCGs (rps16, rpoC1, rpl2, rpl16, ndhA, ndhB, PetB, PetD, and atpF) only possessed one intron (Table 2). The trnK-UUU gene had the largest intron (2532 bp), which included the matK gene. Introns can regulate the gene transcription rate and play a vital role in gene structure and function [22]. Rpl2, which was the only gene with an intron in the ribosomal large subunit of S. divaricata cpDNA, is commonly used as a phylogenetic marker for special species, such as those under tribe Desmodieae [23]. Screening via hybridization demonstrated that the rpl2 intron was lost in at least five other dicotyledon lineages [24]. In higher plants, infA encodes approximately 70 amino acids of the translation initiation factor IF1, which is an important component of protein translation initiation in the organelles [25]. InfA is an extremely active gene during cpDNA evolution and has become repeatedly invalidated in 24 angiosperm lineages, although most angiosperm species seemingly contain the intact gene [26]. Furthermore, infA is considered the most mobile cpDNA gene in plants that has been transferred many times through evolution [27].

Table 1.
List of genes found in the chloroplast genome of the wild S. divaricata.
Table 2.
The genes with introns in the wild S. divaricata chloroplast genome and the length of the exons and introns.
In addition, we analyzed the codon usage preference and relative synonymous codon usage (RSCU) in the cpDNAs of S. divaricata and its related species. Based on the tRNA and PCG sequences, the codon usage frequency in the wild S. divaricata cpDNA was determined (Table S2) and compared to six closely related species, namely, L. seseloides, Libanotis buchtormensis, Seseli montanum, Peucedanum praeruptorum, Angelica paeoniifolia, and Arracacia xanthorrhiza (Figure 3). In total, 24,347 codons were detected in all the coding sequences of S. divaricata. Among these, leucine (Leu) was the most common amino acid, accounting for 10.6% (2573) of the total codons, whereas cysteine (Cys) was the least common (1.0%, 255). The comparison of the GC content in the first to third (GC1–GC3) positions and total GC content (GCs) among the seven cpDNAs indicated that the GC composition of the codons in S. divaricata and L. seseloides cpDNAs was the most similar (Figure 3A–D). Furthermore, the majority of the synonymous codons with RSCU values >1 ended with either adenine (A) or thymine (T) bases (except for TTG and ATG), indicating that codons with A or T ends are common (Table S2, Figure 3E). Notably, Arginine (Arg), Leu, and Serine (Ser) showed a high degree of codon bias among the seven species, whereas tryptophan (Trp) had no codon bias. In addition, we found that the cpDNAs of the wild S. divaricata and the other species preferred TAA as the termination codon. Hypothetically, the best combination of codons can promote the faster and more accurate translation of required proteins. The use of synonymous codons is also influenced by multiple factors, such as genome size, gene length, gene expression level, protein secondary structure, and gene density [28,29]. Therefore, codon preference analysis may be used to examine the balance between mutation preference and natural selection during translation optimization [30].
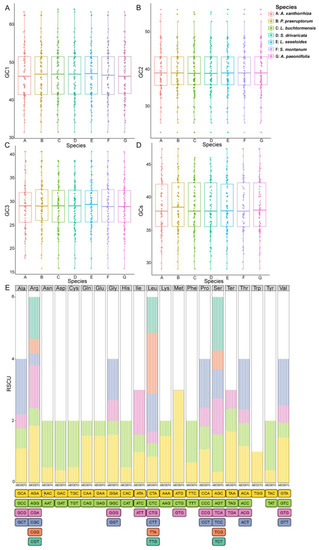
Figure 3.
Comparison of the GC content, codon usage preference, and amino acid proportion in the protein-coding genes of seven chloroplast genomes. (A–D) GC content in the synonymous codons at the first (GC1), second (GC2), and third (GC3) positions and total GC content (GCs). (E) Codon preference and proportion of amino acids based on relative synonymous codon usage (RSCU) values. Ter represents the stop codon. Legend: A, A. xanthorrhiza; B, P. praeruptorum; C, L. buchtormensis; D, S. divaricata; E, L. seseloides; F, S. montanum; G, A. paeoniifolia.
2.2. Comparative CpDNA Analysis of Seven Species under Subfamily Apioideae
The sequence divergence of the cpDNAs among selected species belonging to subfamily Apioideae Drude—L. buchtormensis and S. montanum under tribe Ammineae, P. praeruptorum and A. paeoniifolia under tribe Peucedaneae, A. xanthorrhiza under tribe Selineae, and L. seseloides under tribe Laserpiteae—were examined using the S. divaricata (tribe Laserpiteae) cpDNA as reference (Figure 4). As expected, all cpDNAs exhibited the general structure and order of characteristic genes, with the non-coding regions showing greater variation than the coding regions. Notably, ycf1 (IR and SSC regions) and ycf2 (IR and LSC regions) were quite mutable. Since the lengths of ycf1 and ycf2 located at the boundaries of IR regions are very long, these genes are thus prone to insertion–deletion (InDel), resulting in the considerable differences between the cpDNAs of S. divaricata and the other species. These results indicate that the IR, SSC, and LSC regions rapidly evolved in Apioideae Drude species. Notably, the rRNA sequences were the most conserved among the seven cpDNAs, which is similar to most angiosperms, such as Salvia miltiorrhiza [31] and Phyllostachys sulphurea [32]. We also found that the degree of variation among the IR regions of the seven cpDNAs was low, whereas most of the variation occurred in the SSC regions and in the binding sites of the IR and LSC regions (Figure 2). In addition, all the coding regions in the seven cpDNAs were extracted and evaluated for nucleotide variability. Eight PCGS, namely, rpl32, trnH-GUG, ycf2, ndhI, trnP-UGG, psaJ, psbA, and psaC, possessed the highest Pi values, of which rpl32 was the most variable (Figure 5).
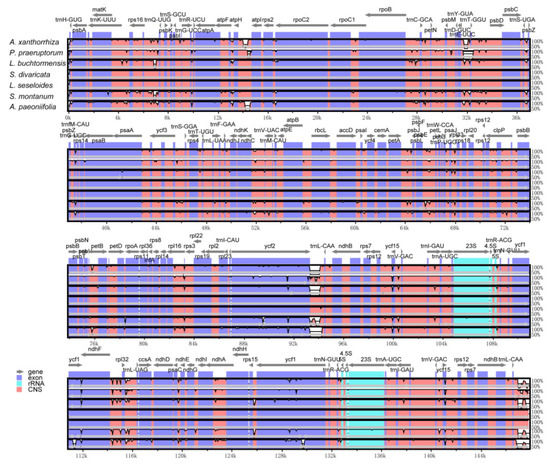
Figure 4.
Comparison of the seven chloroplast genomes belonging to subfamily Apioideae Drude using mVISTA program. Grey arrows and thick black lines above the alignments indicate gene orientations and IR positions, respectively. A cut-off of 70% identity was used for the plots, with the Y-scale representing the percent identity (50–100%). Genome regions are color-coded as protein-coding (exon; blue), ribosomal RNA (rRNA; cyan), and conserved non-coding sequences (CNS; pink).
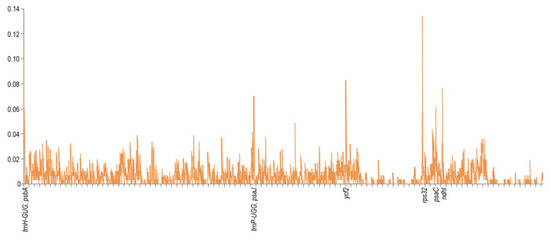
Figure 5.
Comparison of the nucleotide variability (Pi) values among the seven species cp genomes. The Y-axis shows the Pi values; the X-axis shows the genes with high Pi values.
The expansion and contraction at the IR region borders are prevalent in many species and are considered the primary reason for the size differences between plant cpDNAs during evolution [33]. Comparison of the IR/LSC and IR/SSC boundaries in A. xanthorrhiza, P. praeruptorum, S. divaricata, L. seseloides, L. buchtormensis, S. montanum, and A. paeoniifolia was performed to assess the degree of IR expansion or contraction among these species. As expected, S. divaricata and L. seseloides contained similar boundaries in the LSC, SSC, and IR regions, with a small difference in the size of ycf2. This result supports the hypothesis that S. divaricata and L. seseloides are the same species. By contrast, due to the less frequent expansion of ycf2 in the LSC/IRb junction, the IR regions in S. divaricata were much smaller than those in L. buchtormensis and S. montanum. In particular, the ycf2 in the LSC region of S. divaricata showed an 80-bp-long expansion towards the IRb region, whereas those of L. buchtormensis and S. montanum had 1293- and 1302-bp-long expansions towards their IRb regions, respectively. The ndhB/ycf2, ycf1, ndhF, and trnH genes were also found to be located in the LSC/IRb, SSC/IRb, IRa/SSC, and LSC/IRa junctions, respectively (Figure 6). Among these, ycf1, a possible pseudogene located in the IR/SSC boundary, was generated after expansion, which is similar to the corresponding coding gene and can be considered as a non-functional genomic DNA copy. However, ycf1 is not transcribed and has no specific physiological function. The ycf1 sequence exhibited a 1-, 22-, 11-, 11-, 38-, 8-, and 16-bp-long expansion from the IRb to the SSC regions in the cpDNAs of A. xanthorrhiza, P. praeruptorum, S. divaricata, L. seseloides, L. buchtormensis, S. montanum, and A. paeoniifolia, respectively. By contrast, the gaps of trnH sequences in the LSC from the IRa regions of P. praeruptorum, S. divaricata, L. seseloides, L. buchtormensis, and S. montanum were 47, 57, 57, 321, and 663 bp long, respectively. However, A. xanthorrhiza and A. paeoniifolia contained no trnH in the LSC region. Notably, the majority of the cpDNAs contained trnN in the IRa regions, except S. divaricata, L. seseloides, and A. xanthorrhiza, whereas only S. divaricata, L. seseloides, and A. paeoniifolia possessed trnL in the IR regions. Moreover, P. praeruptorum, L. buchtormensis, S. divaricata, and L. seseloides possessed psbA in the LSC regions. Recently, the psbA-trnH intergenic spacer (IGS) region was used as a candidate DNA barcode sequence to identify similar species under the genus Dendrobium [34] and family Umbelliferae [35]. The psbA-trnH IGS can also be used as a barcode to distinguish whether two species belong to the same family [36]. In addition, the trnN in the S. divaricata and L. seseloides cpDNAs may have been lost during recombination. Therefore, we hypothesize that the psbA-trnH IGS can be combined with trnN to develop a DNA barcode for the molecular identification of S. divaricata plants.
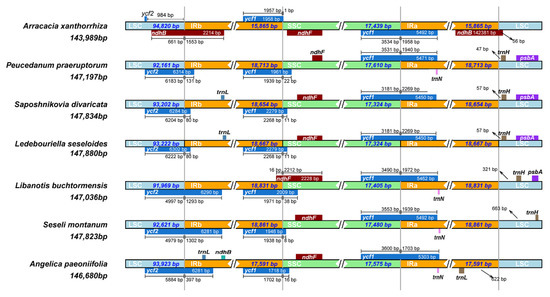
Figure 6.
Comparison of the borders of LSC, SSC, and IR regions among seven cp genomes.
2.3. Identification of Repeat Sequences and SSRs in Wild S. divaricata CpDNA
A total of 33 forward, 14 reverse, and 3 complement repeat sequences were discovered in the wild S. divaricata cpDNA (Table 3). Most of these repeats were between 20 and 50 bp in length. The largest was the 84 bp-long forward repeat in the ycf2 of the LSC region. Notably, LSC was the region with the densest number of repeated sequences. Among these, No. 28–35 were also associated with ycf2, whereas No. 45 was related to ndhA. Ten forward repeats were located in the IR regions, including two repeats (No. 40 and 49) related to ycf15. Moreover, two pairs of repeats (No. 9 and 10) were found to be located in two different regions, specifically in the introns of LSC/SSC and LSC/IRb, respectively.
Table 3.
Repeat sequences in the chloroplast genome of the wild S. divaricata.
SSRs or microsatellites are 1–6 bp repeat sequences commonly distributed throughout the genome. SSRs have been widely employed in studies for species identification, population genetics, and evolutionary history due to their high level of intraspecific polymorphism and uniparental inheritance [37,38]. In total, forty-nine SSRs were discovered in the wild S. divaricata cpDNA, including forty mononucleotide (81.6%), four dinucleotide (8.2%), two trinucleotide (4.1%), and three complex (6.1%) SSRs, most of which were found in the LSC region (Table 4). Furthermore, the three complex SSRs consisted of three mononucleotide and four dinucleotide repeats. A total of 21 SSRs were detected in the genes, and the rest were located in the IGS region. Thirty-three (67.3%) mononucleotide SSRs were mainly composed of short poly A or poly T repeats and rarely contained tandem guanine (G) or cytosine (C) repeats, which corroborate previous reports on other herbs [39]. These SSR markers can be utilized for the conservation study, linkage map construction, and marker-assisted selection of wild S. divaricata and other closely related species.
Table 4.
Simple sequence repeats (SSRs) in the wild S. divaricata chloroplast genome.
2.4. Phylogenetic Analysis of 47 Taxa under Subfamily Apioideae Based on CpDNA Sequences
Based on the successful application of cpDNAs in studying angiosperm phylogeny, complete cpDNA sequences have been widely used to obtain powerful data for developing biosystem models [14]. To study the phylogenetic position of the wild S. divaricata within the Apiaceae subfamily Apioideae Drude, the complete cpDNAs of forty-seven taxa belonging to ten genera under tribes Peucedaneae Drude, Smyrnieae Koch, Ammineae Koch, Laserpiteae Drude, and Selineae Spreng were used for phylogenetic tree construction (Table S3). One species each from tribes Saniculoideae Drude (Sanicula chinensis) and Mackinlayoideae Plunkett and Lowry (Centella asiatica) were selected as outgroups (Figure 7). The maximum likelihood (ML) trees generated using FastTree and IQ-TREE software demonstrated similar results and ensured the reliability of the phylogenetic analysis, but also showed some difference from previous reports [16,17]. Notably, the 100% bootstrap value observed in the clustering of L. seseloides and S. divaricata further supported the hypothesis that the two were the same species. In addition, S. divaricata (Laserpiteae Drude) was discovered to be most closely related to L. buchtormensis from Ammineae Koch, P. japonicum and P. praeruptorum from Peucedaneae Drude, and S. montanum from Ammineae Koch. These results suggest that the genetic relationships between the species under genera Saposhnikovia and Libanotis are closer than those under genera Peucedanum and Seseli, as evidenced by the high bootstrap support values. Furthermore, Laserpiteae Drude and Ammineae Koch species potentially have a closer kinship with each other than with Peucedaneae Drude species, which contradicts the previous reports on cultivated S. divaricata [16,17].
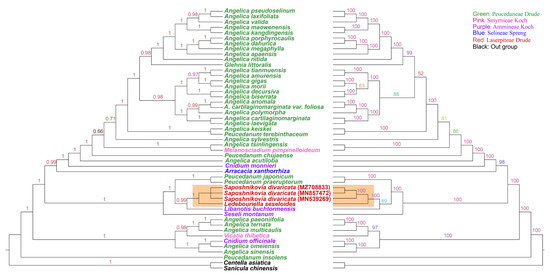
Figure 7.
Phylogeny of 47 taxa within Apioideae Drude species based on the ML analysis of the cp genome’s IRs, LSC, and SSC regions with Sanicula chinensis and Centella asiatica as the outgroups based on FastTree (left) and IQ-TREE (right). The information of all chloroplast genomes used for phylogenetic analysis was shown in Table S3.
3. Materials and Methods
3.1. Sampling, CpDNA Extraction, and Sequencing
Fresh mature leaves were plucked from wild S. divaricata. Total genomic DNA was extracted from young leaves using a Trelief TM Plant Genomic DNA Kit (TsingKe Biotechnology Co., Ltd., Beijing, China). After quality testing, DNA was fragmented and used to set up 350 bp short-insert libraries and the qualified libraries were sequenced with PE 150 bp on the BGISEQ-500 sequencer according to the manufacturer’s instructions. The sequencing depth was 6.0 Gb of 150-bp paired-end reads.
3.2. CpDNA Assembly and Annotation
First, all raw reads were trimmed using Fastp [40]. Subsequently, high-quality reads were mapped to the reference chloroplast genomes of Apioideae obtained from GenBank through Bowtie2 v.2.3.4.3 (Langmead B, et al. https://github.com/BenLangmead/bowtie2, accessed on accessed on 14 September 2020) [41]. The sequence of the coding gene having the maximum coverage was utilized as a seed sequence for de novo assembly by NOVOPlasty v4.2.1 [42]. The assembled cp genomes were annotated with DOGMA [43], GeSeq [44], tRNAscan [45], and ARAGORN [46], then manually adjusted and confirmed using Geneious 9.1.8 (M Kearse, et al. San Diego, CA, https://www.geneious.com/, accessed on accessed on 14 September 2020) [47]. The circular chloroplast genome map was drawn by OrganellarGenomeDRAW tool (OGDRAW) v.1.3.1 (Greiner S, et al. https://chlorobox.mpimp-golm.mpg.de/OGDraw.html, accessed on accessed on 14 September 2020) [48] for further comparison of gene order and content. The other genomes downloaded from GenBank for comparative analysis were re-annotated according to the above method. The assembled cp genome has been deposited to the GenBank with the accession number MZ708833.
3.3. CpDNA Comparison and Sequence Divergence Analysis
The Relative Synonymous Codon Usage (RSCU) values were determined to quantify the extent of the codon usage bias. RSCU was calculated for every codon in each genome according to the published equation [49]. The overall GC content and GC content at the first, second, and third codon positions (GC1, GC2, and GC3, respectively) of the genomes were calculated using EMBOSS software suite [50]. Simple sequence repeats (SSRs) were searched via MISA v1.01 [51] with the following criteria: 10, 6, 5, 5, 5, and 5 repeat units for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides, respectively. Chloroplast genome similarity was assessed using BLAST Atlas on the GView server (Franklin B., et al. https://server.gview.ca/, accessed on 14 September 2020) [52] with S. divaricata genome as a reference. The junction regions between the IR, SSC, and LSC of these plastomes were compared using the IRscope+ online program [53]. The divergent regions were visualized using Shuffle-LAGAN mode [54] included in mVISTA v.2.0 (Frazer K.A., et al., https://genome.lbl.gov/vista/mvista, accessed on 14 September 2020) [55] with S. divaricata genome as a reference. To identify polymorphic regions with substantial variability, the aligned sequences were imported in DnaSP v6.12.03 (DNA Sequences Polymorphism) (Rozas J., et al. http://www.ub.edu/dnasp/, accessed on 14 September 2020) using the sliding window method with a step size of 15 bp and a window length of 200 bp [56].
3.4. Phylogenetic Analysis
The complete cp genomes of forty-seven taxa from the Apiaceae subfamily Apioideae Drude and two species from Saniculoideae Drude (S. chinensis) and Mackinlayoideae Plunkett and Lowry (C. asiatica) as outgroups were employed for the phylogenetic reconstruction. These cpDNAs were downloaded from GenBank in NCBI (Table S3). The whole cpDNA sequence alignment was carried out by using MAFFT v7.450 (Katoh K., et al. https://mafft.cbrc.jp/alignment/software/, accessed on 14 September 2020) [57], and then the regions with consistent site coverage less than 95% were deleted. Maximum likelihood (ML) analysis was performed by FastTree 2.1.11 (Price M.N., et al. http://www.microbesonline.org/fasttree/, accessed on 14 September 2020) [58] and IQ-TREE version 2.1.4 (Minh B.Q., et al. https://github.com/iqtree/iqtree2, accessed on 14 September 2020) [59]. The former was conducted under the best-fit nucleotide substitution model with General Time Reversible + γ (GTR + γ), Shimodaira–Hasegawa test, and the latter was determined using the Akaike Information Criterion (AIC) by ModelFinder in the IQ-TREE package and 1000 bootstrap replicates [60].
4. Conclusions
In this study, we first analyzed the cpDNA of the wild S. divaricata and compared it with its close relatives. The wild S. divaricata cpDNA contained 8 rRNA genes, 36 tRNA genes, and 85 PCGs and had a total GC content of 37.5%. These results are consistent with all the reported cpDNA sequences of S. divaricata and its synonymous species, L. seseloides. Compared to other related species, the non-coding regions exhibited greater variation than the coding regions. The comparison of the IR/LSC and IR/SSC boundaries among seven cpDNAs revealed that the trnN in the wild S. divaricata may have been lost during the reorganization process. Hence, trnN can be combined with the psbA-trnH IGS region as a DNA barcode for the Apioideae Drude species. We also found that the LSC region was a dense region of repeated sequences, in which 49 potentially informative SSRs were identified. Furthermore, the genetic relationship between L. seseloides and S. divaricata was confirmed at the genomic level for the first time. Notably, these two were most closely related to L. buchtormensis, which contradicts previous reports. By contrast, the phylogenetic tree showed that the Laserpiteae Drude and Ammineae Koch species have a close kinship. Overall, our findings contribute important genetic information that may be useful for future studies on the genetic diversity and phylogenetic relationships of the Apioideae species.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/genes13050931/s1, Table S1: Base composition in the chloroplast genome of the wild S. divaricata. Table S2: All chloroplast genome information of S. divaricata and its synonyms. Table S3: Codon-anticodon recognition patterns and codon usage for the wild S. divaricata chloroplast genome. Table S4: The information of all complete chloroplast genomes of Apioideae Drude used for phylogenetic analysis in this study.
Author Contributions
Conceptualization, B.H. and D.L.; methodology, S.Y.; software, M.L.; validation, G.W. and W.W.; formal analysis, H.L. and C.C.; investigation, W.W.; resources, T.X.; data curation, M.L.; writing—original draft preparation, S.Y. and F.G.; writing—review and editing, B.H. and D.L.; visualization, W.W. and H.L.; supervision, B.H., D.L. and S.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the China Agriculture Research System of MOF and MARA, Major Program of Increase or Decrease in The Central Government (2060302), High level talent project of West Anhui University (WGKQ2021024), and Key projects of excellent young talents support program of Anhui universities (gxyqZD2020040).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are openly available in GeneBank.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kreiner, J.; Pang, E.; Lenon, G.B.; Yang, A.W.H. Saposhnikoviae divaricata: A phytochemical, pharmacological, and pharmacokinetic review. Chin. J. Nat. Med. 2017, 15, 255–264. [Google Scholar] [CrossRef]
- Yang, J.L.; Dhodary, B.; Ha, T.K.Q.; Kim, J.; Kim, E.; Oh, W.K. Three new coumarins from Saposhnikovia divaricata and their porcine epidemic diarrhea virus (PEDV) inhibitory activity. Tetrahedron 2015, 71, 4651–4658. [Google Scholar] [CrossRef] [PubMed]
- Batsukh, Z.; Toume, K.; Javzan, B.; Kazuma, K.; Cai, S.; Hayashi, S.; Atsumi, T.; Yoshitomi, T.; Uchiyama, N.; Maruyama, T.; et al. Characterization of metabolites in Saposhnikovia divaricata root from Mongolia. J. Nat. med. 2020, 75, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wang, C.C.; Wang, W.L.; Xu, J.P.; Wang, J.; Zhang, C.H.; Li, M.H. Saposhnikovia divaricata—An ethnopharmacological, phytochemical and pharmacological review. Chin. J. Integr. Med. 2020, 26, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Tai, J.; Cheung, S. Anti-proliferative and antioxidant activities of Saposhnikovia divaricata. Oncol. Rep. 2007, 18, 227–234. [Google Scholar] [CrossRef][Green Version]
- Chun, J.M.; Kim, H.S.; Lee, A.Y.; Kim, S.H.; Kim, H.K. Anti-inflammatory and antiosteoarthritis effects of Saposhnikovia divaricata ethanol extract: In vitro and in vivo studies. Evid. Based Complementary Altern. Med. 2016, 2016, 1984238. [Google Scholar]
- Okuyama, E.; Hasegawa, T.; Matsushita, T.; Fujimoto, H.; Ishibashi, M.; Yamazaki, M. Analgesic components of saposhnikovia root (Saposhnikovia divaricata). Chem. Pharm. Bull. 2001, 49, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Niu, Y.; Zheng, J.; Liu, H.; Jiang, G.; Chen, J.; Hong, M. Radix Saposhnikovia extract suppresses mouse allergic contact dermatitis by regulating dendritic-cell-activated Th1 cells. Phytomedicine 2015, 22, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, H.; Emes, M. Nonphotosynthetic metabolism in plastids. Annu. Rev. Plant Biol. 2000, 51, 111–140. [Google Scholar] [CrossRef] [PubMed]
- Vesteg, M.; Vacula, R.; Krajcovic, J. On the origin of chloroplasts, import mechanisms of chloroplast-targeted proteins, and loss of photosynthetic ability–review. Folia Microbiol. 2009, 54, 303–321. [Google Scholar] [CrossRef]
- Henry, R.J. Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants; CABI Publishing: Cambridge, MA, USA, 2005. [Google Scholar]
- Raubeson, L.A.; Jansen, R.K. Chloroplast genomes of plants. In Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants; CABI Publishing: Cambridge, MA, USA, 2005; pp. 45–68. [Google Scholar]
- Whitfeld, P.R.; Bottomley, W. Organization and structure of chloroplast genes. Annu. Rev. Plant Physiol. 1983, 34, 279–310. [Google Scholar] [CrossRef]
- Jian, H.Y.; Zhang, Y.H.; Yan, H.J.; Qiu, X.Q.; Wang, Q.G.; Li, S.B.; Zhang, S.D. The complete chloroplast genome of a key ancestor of modern roses, Rosa chinensis var. spontanea, and a comparison with congeneric species. Molecules 2018, 23, 389. [Google Scholar] [CrossRef] [PubMed]
- Saina, J.K.; Li, Z.Z.; Gichira, A.W.; Liao, Y.Y. The complete chloroplast genome sequence of tree of heaven (Ailanthus altissima (Mill.) (Sapindales: Simaroubaceae), an important pantropical tree. Int. J. Mol. Sci. 2018, 19, 929. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Zhu, Z.; Zhang, H.; Zhong, Y.; Wang, W.; Zhang, J.; Wu, J. The complete chloroplast genome of Saposhnikovia divaricata. Mitochondrial DNA B 2019, 5, 360–361. [Google Scholar] [CrossRef]
- Li, L.; Geng, M.; Li, Y.; Xu, Z.; Xu, M.; Li, M. Characterization of the complete plastome of Saposhnikovia divaricata (Turcz.) Schischk. Mitochondrial DNA B 2020, 5, 786–787. [Google Scholar] [CrossRef]
- Lee, H.O.; Kim, K.; Lee, S.C.; Lee, J.; Lee, J.; Kim, S.; Yang, T.J. The complete chloroplast genome sequence of Ledebouriella seseloides (Hoffm.) H. Wolff. Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 3498–3499. [Google Scholar] [CrossRef]
- Komarov, V.L. Saposhnikovia divaricata (Turcz.) Schischk. 1951. Available online: https://www.ipni.org/n/847902-1 (accessed on 14 September 2020).
- Zhang, R.; Li, Q.; Gao, J.; Qu, M.; Ding, P. The complete chloroplast genome sequence of the medicinal plant Morinda officinalis (Rubiaceae), an endemic to China. Mitochondrial DNA A 2016, 27, 4324–4325. [Google Scholar] [CrossRef]
- Ding, P.; Shao, Y.; Li, Q.; Gao, J.; Zhang, R.; Lai, X.; Wang, D.; Zhang, H. The complete chloroplast genome sequence of the medicinal plant Andrographis paniculata. Mitochondrial DNA A 2016, 27, 2347–2348. [Google Scholar] [CrossRef]
- Callis, J.; Fromm, M.; Walbot, V. Introns increase gene expression in cultured maize cells. Genes Dev. 1987, 1, 1183–1200. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.D.; Doyle, J.J. The chloroplast rpl2 intron and ORF184 as phylogenetic markers in the legume tribe Desmodieae. Syst. Bot. 1997, 22, 133–138. [Google Scholar] [CrossRef]
- Downie, S.R.; Olmstead, R.G.; Zurawski, G.; Soltis, D.E.; Soltis, P.S.; Watson, J.C.; Palmer, J.D. Six independent losses of the chloroplast DNA rpl2 intron in dicotyledons: Molecular and phylogenetic implications. Evolution 1991, 45, 1245–1259. [Google Scholar] [CrossRef] [PubMed]
- Hägg, P.; Pohl, J.W.; Abdulkarim, F.; Isaksson, L.A. A host/plasmid system that is not dependent on antibiotics and antibiotic resistance genes for stable plasmid maintenance in Escherichia coli. J. Biotechnol. 2004, 111, 17–30. [Google Scholar] [CrossRef]
- Millen, R.S.; Olmstead, R.G.; Adams, K.L.; Palmer, J.D.; Lao, N.T.; Heggie, L.; Kavanagh, T.A.; Hibberd, J.M.; Gray, J.C.; Morden, C.W.; et al. Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 2001, 13, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, K.H.; Morden, C.W.; Ems, S.C.; Palmer, J.D. Rapid evolution of the plastid translational apparatus in a nonphotosynthetic plant: Loss or accelerated sequence evolution of tRNA and ribosomal protein genes. J. Mol. Evol. 1992, 35, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Presnyak, V.; Alhusaini, N.; Chen, Y.H.; Martin, S.; Morris, N.; Kline, N.; Olson, S.; Weinberg, D.; Baker, K.E.; Graveley, B.R.; et al. Codon optimality is a major determinant of mRNA stability. Cell 2015, 160, 1111–1124. [Google Scholar] [CrossRef]
- Hanson, G.; Coller, J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 2018, 19, 20. [Google Scholar] [CrossRef]
- Coghlan, A.; Wolfe, K.H. Relationship of codon bias to mRNA concentration and protein length in Saccharomyces cerevisiae. Yeast 2000, 16, 1131–1145. [Google Scholar] [CrossRef]
- Qian, J.; Song, J.; Gao, H.; Zhu, Y.; Xu, J.; Pang, X.; Yao, H.; Sun, C.; Li, X.; Li, C.; et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 2013, 8, e57607. [Google Scholar] [CrossRef]
- Wang, L.; Yu, X.; Xu, W.; Zhang, J.; Lin, H.; Zhao, Y. Complete chloroplast genome sequencing support Angelica decursiva is an independent species from Peucedanum praeruptorum. Physiol. Mol. Biol. Plants 2021, 27, 2503–2515. [Google Scholar] [CrossRef]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Wu, C.L.; Su, T.M.; Chaw, S.M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef]
- Yao, H.; Song, J.Y.; Ma, X.Y.; Liu, C.; Li, Y.; Xu, H.X.; Han, J.P.; Duan, L.S.; Chen, S.L. Identification of Dendrobium species by a candidate DNA barcode sequence: The chloroplast psbA-trnH intergenic region. Planta Med. 2009, 75, 667–669. [Google Scholar] [CrossRef] [PubMed]
- Degtjareva, G.V.; Logacheva, M.D.; Samigullin, T.H.; Terentieva, E.I.; Valiejo-Roman, C.M. Organization of chloroplast psbA-trnH intergenic spacer in dicotyledonous angiosperms of the family Umbelliferae. Biochemistry 2012, 77, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Aldrich, J.; Cherney, B.W.; Merlin, E. The role of insertions/deletions in the evolution of the intergenic region between psbA and trnH in the chloroplast genome. Curr. Genet. 1988, 14, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Zalapa, J.E.; Cuevas, H.; Zhu, H.; Steffan, S.; Senalik, D.; Zeldin, E.; McCown, B.; Harbut, R.; Simon, P. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am. J. Bot. 2012, 99, 193–208. [Google Scholar] [CrossRef]
- Cato, S.; Richardson, T. Inter-and intraspecific polymorphism at chloroplast SSR loci and the inheritance of plastids in Pinus radiata D. Don. Theor. Appl. Genet. 1996, 93, 587–592. [Google Scholar] [CrossRef]
- Liu, Y.; Huo, N.; Dong, L.; Wang, Y.; Zhang, S.; Young, H.A.; Feng, X.; Gu, Y.Q. Complete chloroplast genome sequences of Mongolia medicine Artemisia frigida and phylogenetic relationships with other plants. PLoS ONE 2013, 8, e57533. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-fast All-In-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods. 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De Novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq-Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [PubMed]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) Version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.H. The codon Adaptation Indexa measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The european molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Petkau, A.; Stuart-Edwards, M.; Stothard, P.; Van Domselaar, G. Interactive microbial genome visualization with GView. Bioinformatics 2010, 26, 3125–3126. [Google Scholar] [CrossRef]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An Online Program to Visualize the junction Sites of Chloroplast Genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Brudno, M.; Do, C.B.; Cooper, G.M.; Kim, M.F.; Davydov, E.; NISC Comparative Sequencing Program; Green, E.D.; Sidow, A.; Batzoglou, S. LAGAN and Multi-LAGAN: Efficient Tools for Large-Scale Multiple Alignment of Genomic DNA. Genome Res. 2003, 13, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational Tools for Comparative Genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective stochastic algorithm for estimating Maximum-Likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017, 14, 587–589. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).