
Mobilome of Apicomplexa Parasites



Abstract
1. Introduction
2. Materials and Methods
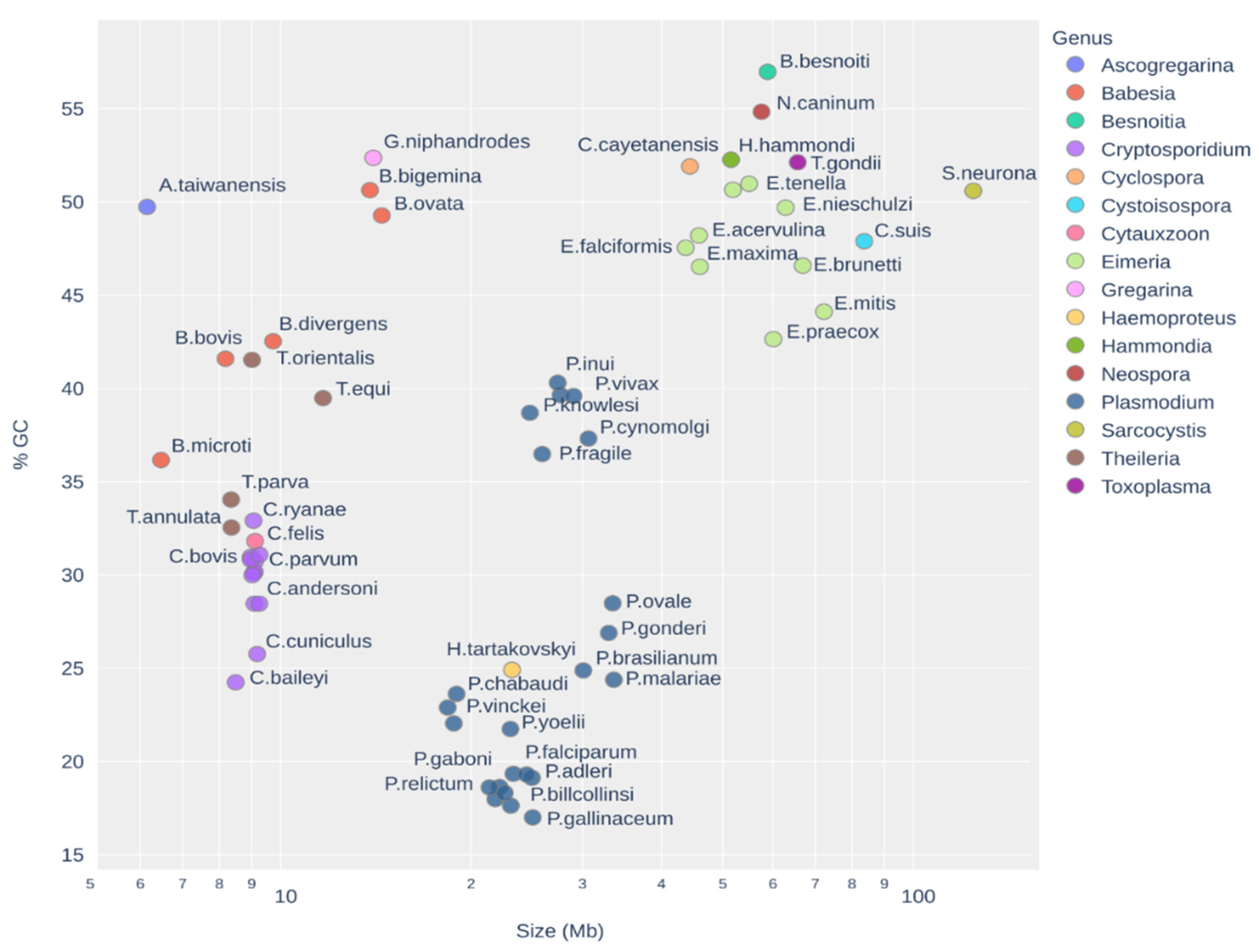
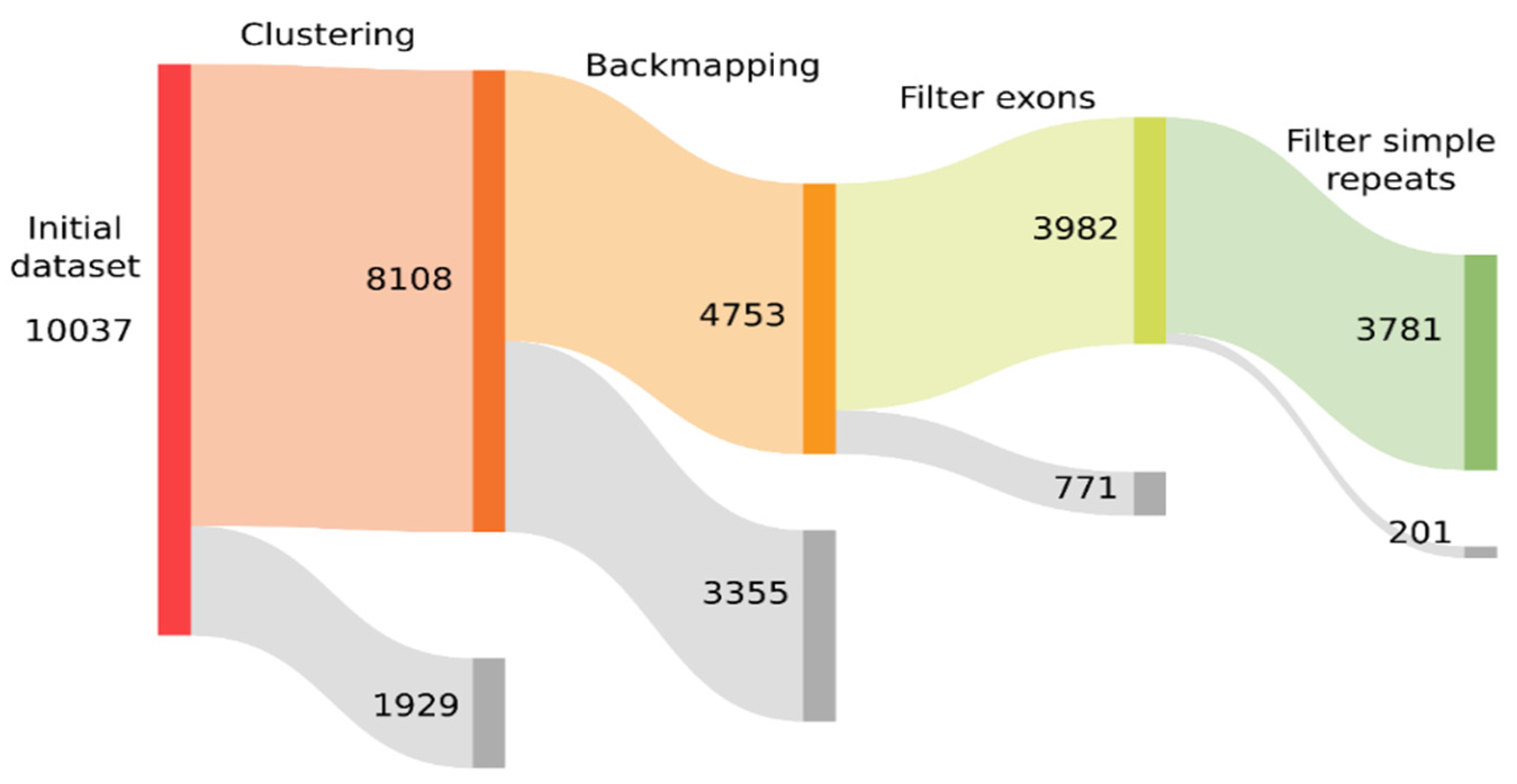
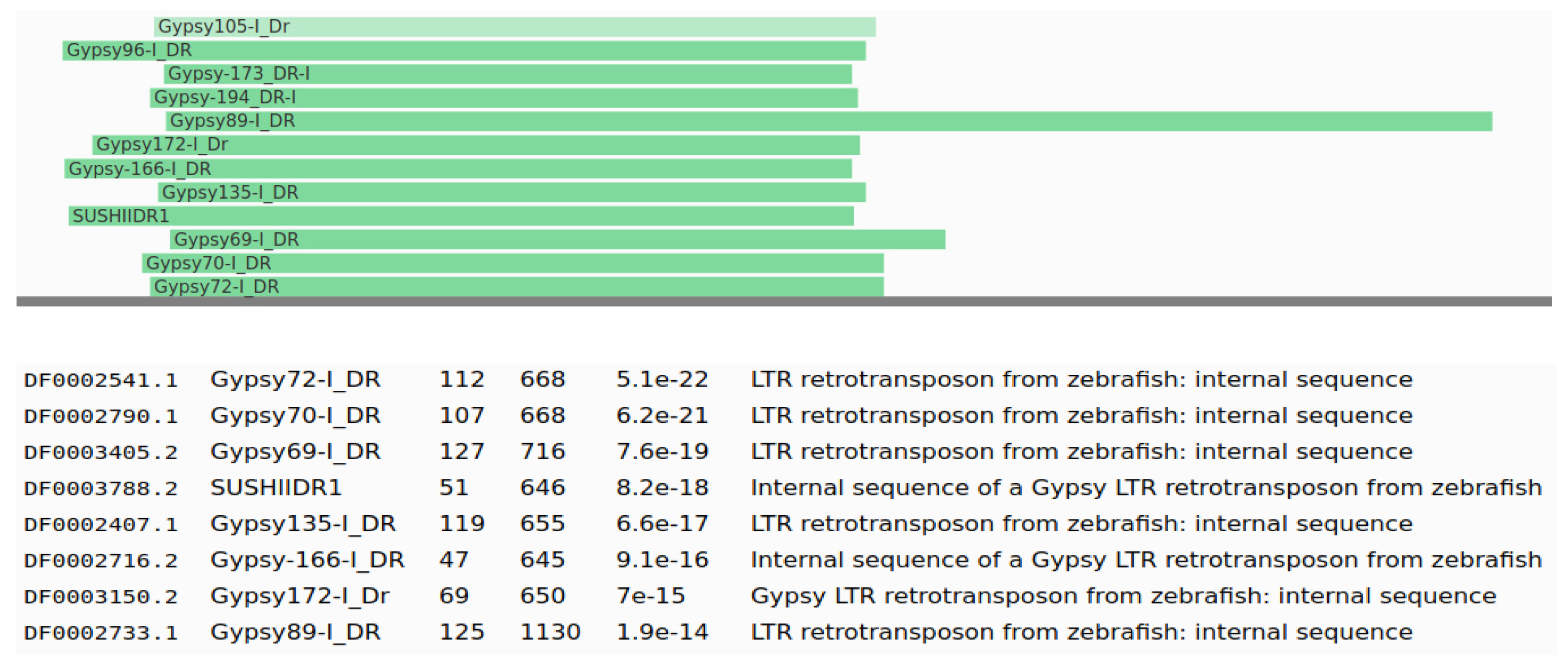
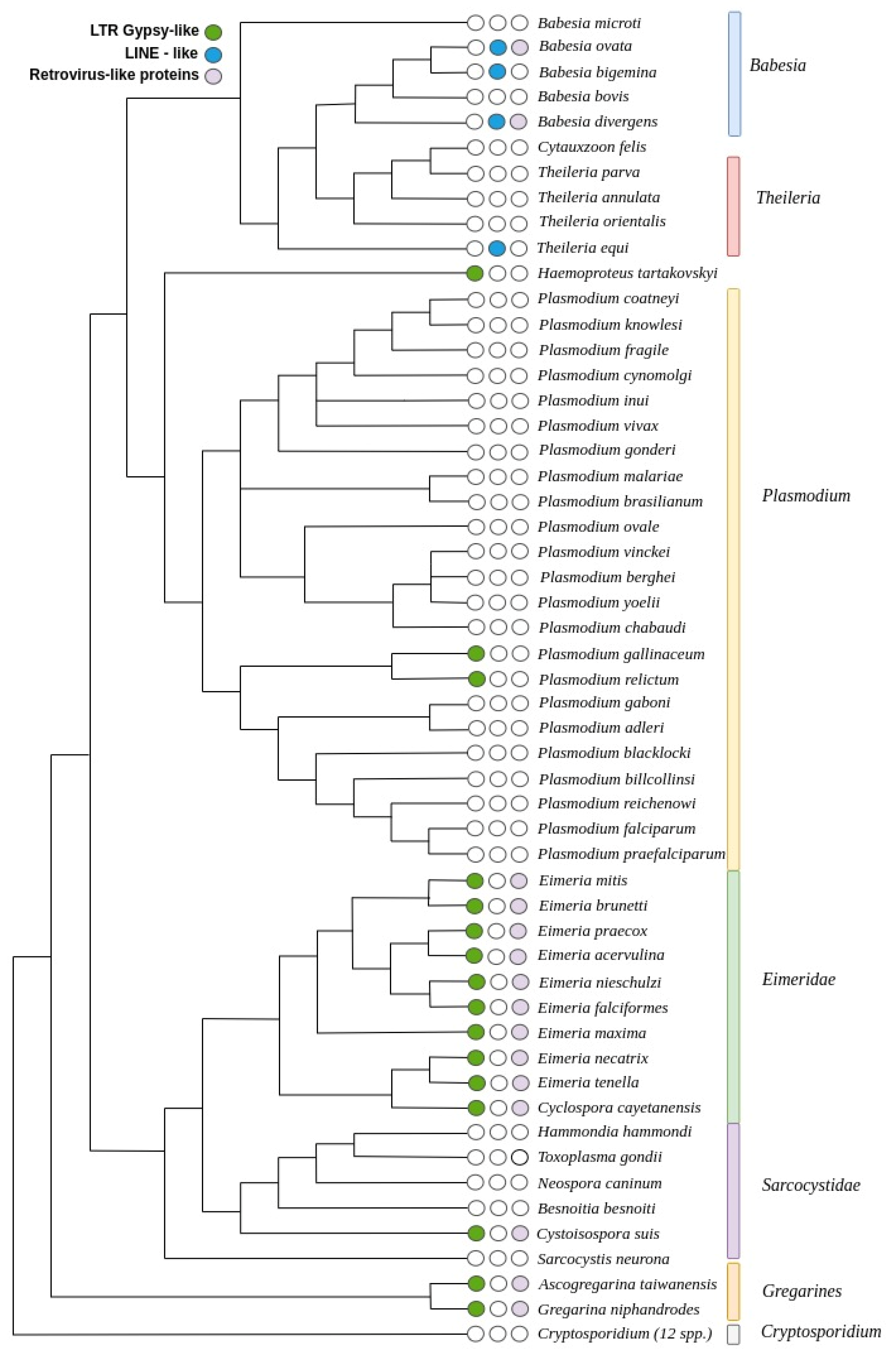
3. Results
3.1. Building and Filtering TE Models
3.2. Identification and Classification of TEs
3.3. Distribution of TEs on the Apicomplexa Phylogeny
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Makałowski, W. Genomic Scrap Yard: How Genomes Utilize All That Junk. Gene 2000, 259, 61–67. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Bakre, A.; Bhattacharya, A. Mobile Genetic Elements in Protozoan Parasites. J. Genet. 2002, 81, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Levine, N.D. Phylum II. Apicomplexa. In An Illustrated Guide to the Protozoa; Lee, J.J., Hunter, S.H., Bovee, E.C., Eds.; Society of Protozoologists: Lawrence, KS, USA, 1985; ISBN 978-0-935868-13-5. [Google Scholar]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a Database of Repetitive Elements in Eukaryotic Genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Durand, P.M.; Oelofse, A.J.; Coetzer, T.L. An Analysis of Mobile Genetic Elements in Three Plasmodium Species and Their Potential Impact on the Nucleotide Composition of the P. Falciparum Genome. BMC Genom. 2006, 7, 282. [Google Scholar] [CrossRef]
- Böhme, U.; Otto, T.D.; Cotton, J.A.; Steinbiss, S.; Sanders, M.; Oyola, S.O.; Nicot, A.; Gandon, S.; Patra, K.P.; Herd, C.; et al. Complete Avian Malaria Parasite Genomes Reveal Features Associated with Lineage-Specific Evolution in Birds and Mammals. Genome Res. 2018, 28, 547–560. [Google Scholar] [CrossRef]
- Templeton, T.J.; Enomoto, S.; Chen, W.-J.; Huang, C.-G.; Lancto, C.A.; Abrahamsen, M.S.; Zhu, G. A Genome-Sequence Survey for Ascogregarina Taiwanensis Supports Evolutionary Affiliation but Metabolic Diversity between a Gregarine and Cryptosporidium. Mol. Biol. Evol. 2010, 27, 235–248. [Google Scholar] [CrossRef]
- Ling, K.-H.; Rajandream, M.-A.; Rivailler, P.; Ivens, A.; Yap, S.-J.; Madeira, A.M.B.N.; Mungall, K.; Billington, K.; Yee, W.-Y.; Bankier, A.T.; et al. Sequencing and Analysis of Chromosome 1 of Eimeria Tenella Reveals a Unique Segmental Organization. Genome Res. 2007, 17, 311–319. [Google Scholar] [CrossRef][Green Version]
- Reid, A.J.; Blake, D.P.; Ansari, H.R.; Billington, K.; Browne, H.P.; Bryant, J.; Dunn, M.; Hung, S.S.; Kawahara, F.; Miranda-Saavedra, D.; et al. Genomic Analysis of the Causative Agents of Coccidiosis in Domestic Chickens. Genome Res. 2014, 24, 1676–1685. [Google Scholar] [CrossRef]
- Mathur, V.; Kolísko, M.; Hehenberger, E.; Irwin, N.A.T.; Leander, B.S.; Kristmundsson, Á.; Freeman, M.A.; Keeling, P.J. Multiple Independent Origins of Apicomplexan-Like Parasites. Curr. Biol. 2019, 29, 2936–2941.e5. [Google Scholar] [CrossRef]
- DeBarry, J.D.; Kissinger, J.C. Jumbled Genomes: Missing Apicomplexan Synteny. Mol. Biol. Evol. 2011, 28, 2855–2871. [Google Scholar] [CrossRef]
- Wasmuth, J.; Daub, J.; Peregrín-Alvarez, J.M.; Finney, C.A.M.; Parkinson, J. The Origins of Apicomplexan Sequence Innovation. Genome Res. 2009, 19, 1202–1213. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; Mcveigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A Comprehensive Update on Curation, Resources and Tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.; Hubley, R. RepeatModeler Open-1.0; 2008. Available online: https://repeatmasker.org/RepeatModeler (accessed on 5 April 2022).
- Rodriguez, M.; Makalowski, W. Software Evaluation for de Novo Detection of Transposons. Mobile DNA 2022, 13, 14. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Smit, A.; Hubley, R.; Green, P. RepeatMasker Open-4.0; 2013. Available online: http://www.repeatmasker.org (accessed on 5 April 2022).
- Benson, G. Tandem Repeats Finder: A Program to Analyze DNA Sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. TRNAscan-SE 2.0: Improved Detection and Functional Classification of Transfer RNA Genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Drost, H.-G. LTRpred: De Novo Annotation of Intact Retrotransposons. J. Open Source Softw. 2020, 5, 2170. [Google Scholar] [CrossRef]
- Rho, M.; Tang, H. MGEScan-Non-LTR: Computational Identification and Classification of Autonomous Non-LTR Retrotransposons in Eukaryotic Genomes. Nucleic Acids Res. 2009, 37, e143. [Google Scholar] [CrossRef]
- Hubley, R.; Finn, R.D.; Clements, J.; Eddy, S.R.; Jones, T.A.; Bao, W.; Smit, A.F.A.; Wheeler, T.J. The Dfam Database of Repetitive DNA Families. Nucleic Acids Res. 2016, 44, D81–D89. [Google Scholar] [CrossRef]
- HMMER. Available online: http://hmmer.org (accessed on 5 April 2022).
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Gish, W. WU BLAST. 1996–2019. Available online: https://blast.advbiocomp.com (accessed on 5 April 2022).
- Abrusán, G.; Grundmann, N.; DeMester, L.; Makalowski, W. TEclass—A Tool for Automated Classification of Unknown Eukaryotic Transposable Elements. Bioinformatics 2009, 25, 1329–1330. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.J. Large, Rapidly Evolving Gene Families Are at the Forefront of Host-Parasite Interactions in Apicomplexa. Parasitology 2015, 142 (Suppl. 1), S57–S70. [Google Scholar] [CrossRef] [PubMed]
- Carreno, R.A.; Martin, D.S.; Barta, J.R. Cryptosporidium Is More Closely Related to the Gregarines than to Coccidia as Shown by Phylogenetic Analysis of Apicomplexan Parasites Inferred Using Small-Subunit Ribosomal RNA Gene Sequences. Parasitol. Res. 1999, 85, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Cavalier-Smith, T. Gregarine Site-Heterogeneous 18S RDNA Trees, Revision of Gregarine Higher Classification, and the Evolutionary Diversification of Sporozoa. Eur. J. Protistol. 2014, 50, 472–495. [Google Scholar] [CrossRef] [PubMed]
- Cornillot, E.; Hadj-Kaddour, K.; Dassouli, A.; Noel, B.; Ranwez, V.; Vacherie, B.; Augagneur, Y.; Brès, V.; Duclos, A.; Randazzo, S.; et al. Sequencing of the Smallest Apicomplexan Genome from the Human Pathogen Babesia Microti. Nucleic Acids Res. 2012, 40, 9102–9114. [Google Scholar] [CrossRef]
- Arisue, N.; Hashimoto, T.; Mitsui, H.; Palacpac, N.M.Q.; Kaneko, A.; Kawai, S.; Hasegawa, M.; Tanabe, K.; Horii, T. The Plasmodium Apicoplast Genome: Conserved Structure and Close Relationship of P. Ovale to Rodent Malaria Parasites. Mol. Biol. Evol. 2012, 29, 2095–2099. [Google Scholar] [CrossRef]
- Janouškovec, J.; Paskerova, G.G.; Miroliubova, T.S.; Mikhailov, K.V.; Birley, T.; Aleoshin, V.V.; Simdyanov, T.G. Apicomplexan-like Parasites Are Polyphyletic and Widely but Selectively Dependent on Cryptic Plastid Organelles. eLife 2019, 8, e49662. [Google Scholar] [CrossRef]
- Heitlinger, E.; Spork, S.; Lucius, R.; Dieterich, C. The Genome of Eimeria Falciformis—Reduction and Specialization in a Single Host Apicomplexan Parasite. BMC Genom. 2014, 15, 696. [Google Scholar] [CrossRef]
- Palmieri, N.; Shrestha, A.; Ruttkowski, B.; Beck, T.; Vogl, C.; Tomley, F.; Blake, D.P.; Joachim, A. The Genome of the Protozoan Parasite Cystoisospora Suis and a Reverse Vaccinology Approach to Identify Vaccine Candidates. Int. J. Parasitol. 2017, 47, 189–202. [Google Scholar] [CrossRef]
- Liu, S.; Wang, L.; Zheng, H.; Xu, Z.; Roellig, D.M.; Li, N.; Frace, M.A.; Tang, K.; Arrowood, M.J.; Moss, D.M.; et al. Comparative Genomics Reveals Cyclospora Cayetanensis Possesses Coccidia-like Metabolism and Invasion Components but Unique Surface Antigens. BMC Genom. 2016, 17, 316. [Google Scholar] [CrossRef]
- Blazejewski, T.; Nursimulu, N.; Pszenny, V.; Dangoudoubiyam, S.; Namasivayam, S.; Chiasson, M.A.; Chessman, K.; Tonkin, M.; Swapna, L.S.; Hung, S.S.; et al. Systems-Based Analysis of the Sarcocystis Neurona Genome Identifies Pathways That Contribute to a Heteroxenous Life Cycle. mBio 2015, 6, e02445-14. [Google Scholar] [CrossRef] [PubMed]
- Ågren, J.A.; Wright, S.I. Co-Evolution between Transposable Elements and Their Hosts: A Major Factor in Genome Size Evolution? Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol. 2011, 19, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Keeling, P.J. Reduction and Compaction in the Genome of the Apicomplexan Parasite Cryptosporidium Parvum. Dev. Cell 2004, 6, 614–616. [Google Scholar] [CrossRef]
- Abrahamsen, M.S.; Templeton, T.J.; Enomoto, S.; Abrahante, J.E.; Zhu, G.; Lancto, C.A.; Deng, M.; Liu, C.; Widmer, G.; Tzipori, S.; et al. Complete Genome Sequence of the Apicomplexan, Cryptosporidium Parvum. Science 2004, 304, 441–445. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Number of Species |
|---|---|
| Ascogregarina | 1 |
| Babesia | 5 |
| Besnoitia | 1 |
| Cryptosporidium | 12 |
| Cyclospora | 1 |
| Cystoisospora | 1 |
| Cytauxzoon | 1 |
| Eimeria | 9 |
| Gregarina | 1 |
| Hammondia | 1 |
| Haemoproteus | 1 |
| Neospora | 1 |
| Plasmodium | 23 |
| Sarcocystis | 1 |
| Theileria | 4 |
| Toxoplasma | 1 |
| Species | Assembly Genome Length (nt) | TE Coverage (nt) | % of Genome with TEs | Main TE Family |
|---|---|---|---|---|
| Ascogregarina taiwanensis | 6,149,411 | 97,453 | 1.58 | LTR Gypsy-like |
| Babesia bigemina | 13,840,936 | 133,787 | 0.97 | LINE-like |
| Babesia divergens | 9,725,408 | 41,125 | 0.42 | LINE-like |
| Babesia ovata | 14,453,397 | 77,825 | 0.54 | LINE-like |
| Cyclospora cayetanensis | 44,363,576 | 2,176,795 | 4.91 | LTR Gypsy-like |
| Cystoisospora suis | 83,637,532 | 1,169,252 | 1.40 | LTR Gypsy-like |
| Eimeria acervulina | 45,830,609 | 783,904 | 1.71 | LTR Gypsy-like |
| Eimeria brunetti | 66,890,165 | 1,567,917 | 2.34 | LTR Gypsy-like |
| Eimeria falciformis | 43,671,268 | 256,248 | 0.59 | LTR Gypsy-like |
| Eimeria maxima | 45,975,062 | 470,783 | 1.02 | LTR Gypsy-like |
| Eimeria mitis | 72,240,319 | 1,298,206 | 1.80 | LTR Gypsy-like |
| Eimeria necatrix | 55,007,932 | 920,585 | 1.67 | LTR Gypsy-like |
| Eimeria nieschulzi | 62,832,469 | 2,365,007 | 3.76 | LTR Gypsy-like |
| Eimeria praecox | 60,083,328 | 1,423,429 | 2.37 | LTR Gypsy-like |
| Eimeria tenella | 51,859,607 | 637,259 | 1.23 | LTR Gypsy-like |
| Gregaina niphandrodes | 14,009,070 | 432,988 | 3.09 | LTR Gypsy-like |
| Haemoproteus tartakovskyi | 23,209,007 | 12,683 | 0.05 | LTR Gypsy-like |
| Plasmodium gallinaceum | 25,034,007 | 1,351,686 | 5.40 | LTR Gypsy-like |
| Plasmodium relictum | 22,607,426 | 1,105,627 | 4.89 | LTR Gypsy-like |
| Theileria equi | 11,674,479 | 101,054 | 0.87 | LINE-like |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez, M.; Makalowski, W. Mobilome of Apicomplexa Parasites. Genes 2022, 13, 887. https://doi.org/10.3390/genes13050887
Rodriguez M, Makalowski W. Mobilome of Apicomplexa Parasites. Genes. 2022; 13(5):887. https://doi.org/10.3390/genes13050887
Chicago/Turabian StyleRodriguez, Matias, and Wojciech Makalowski. 2022. "Mobilome of Apicomplexa Parasites" Genes 13, no. 5: 887. https://doi.org/10.3390/genes13050887
APA StyleRodriguez, M., & Makalowski, W. (2022). Mobilome of Apicomplexa Parasites. Genes, 13(5), 887. https://doi.org/10.3390/genes13050887