
A New Computational Deconvolution Algorithm for the Analysis of Forensic DNA Mixtures with SNP Markers


Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples, SNP Typing, and Data Description
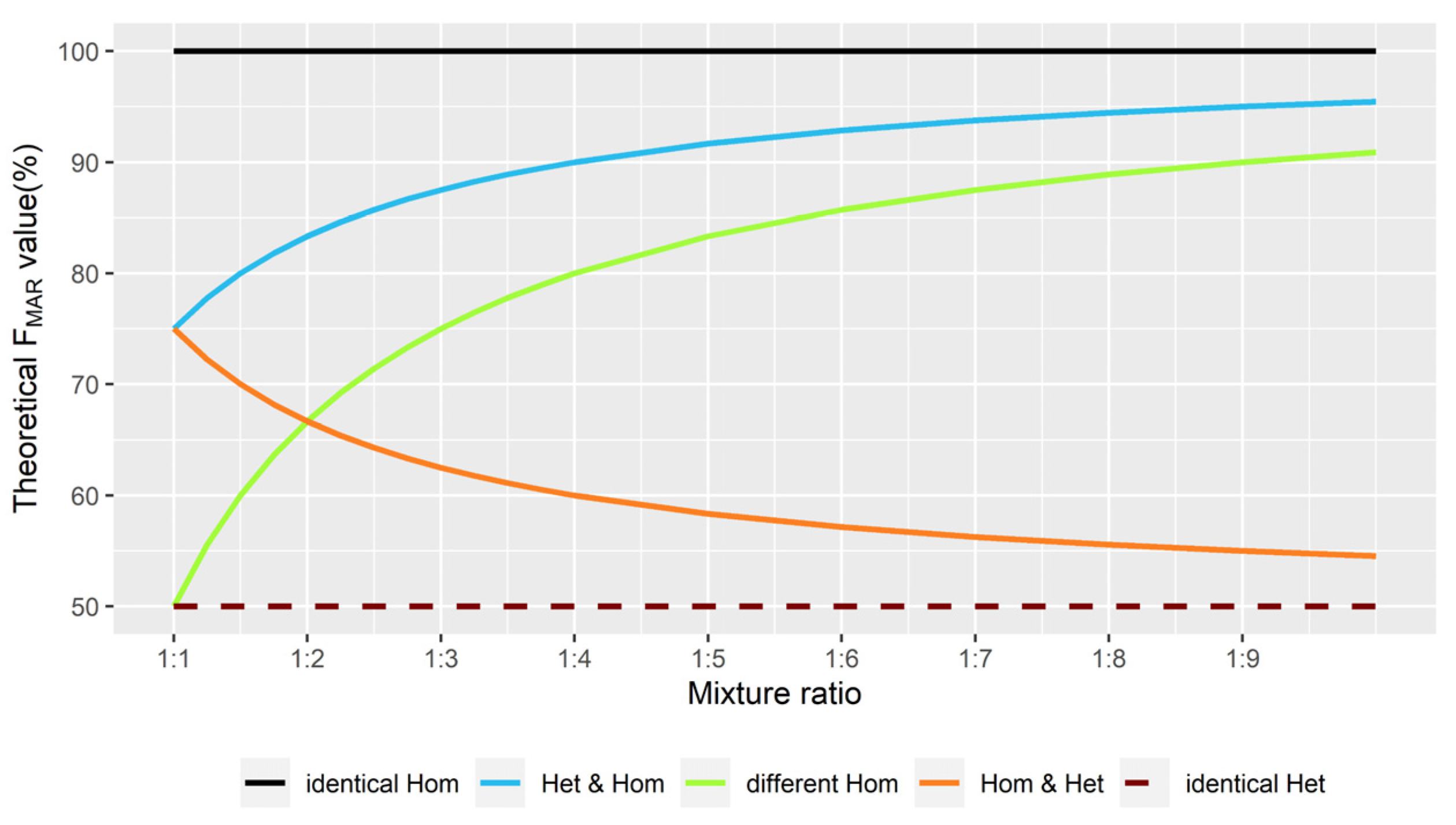
2.2. Theoretical FMAR Values at Different Mixture Ratios
2.3. Simulations with In Silico Mixtures
2.3.1. Splitting BAM Files
2.3.2. Extracting and Merging
2.3.3. Generating CSV Files and Evaluating Simulations
2.4. Clustering FMAR with K-Means Method
2.4.1. A-SNP
2.4.2. Y-SNP
2.5. Validation with In Vitro Mixtures
3. Results and Discussion
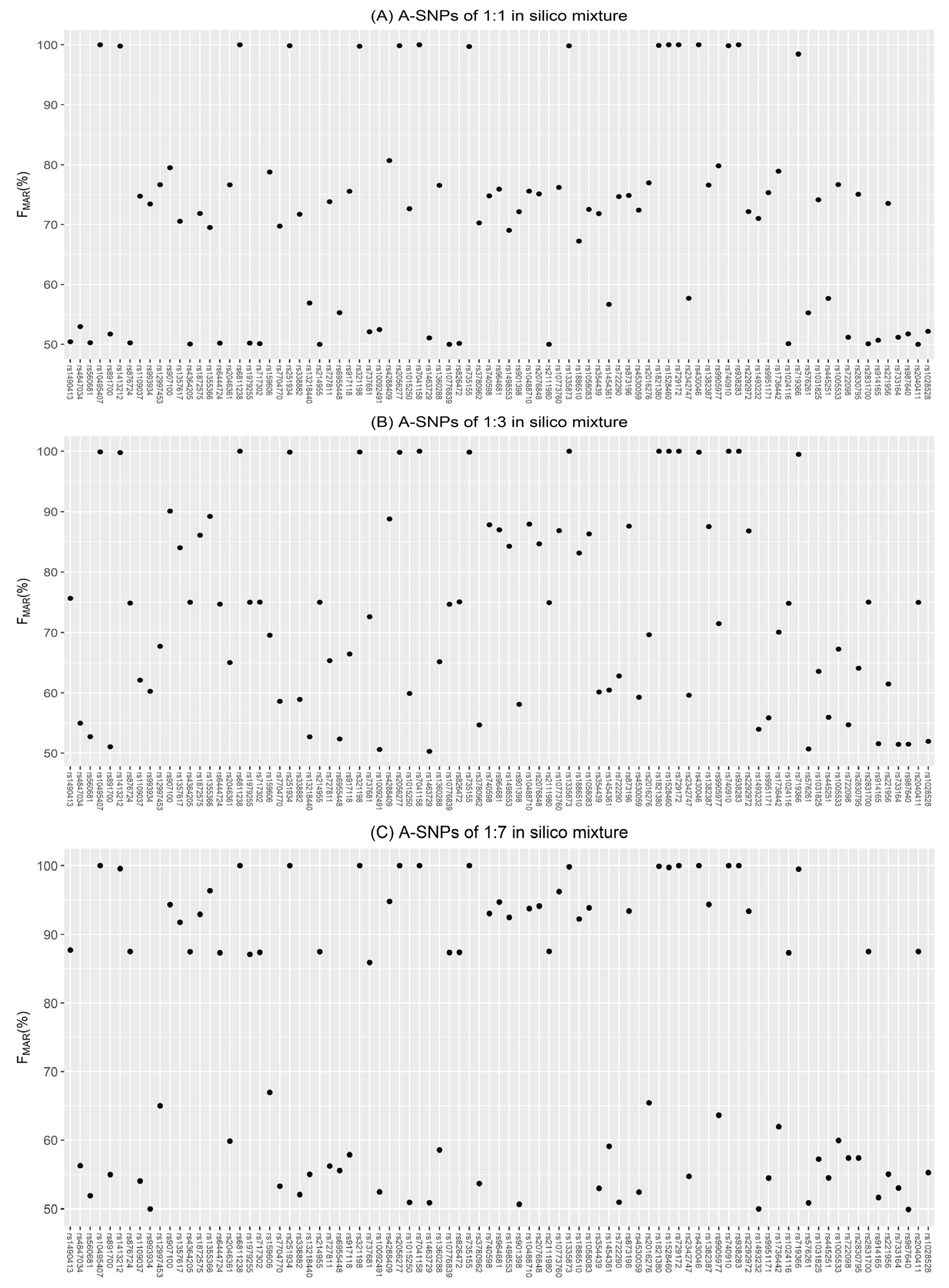
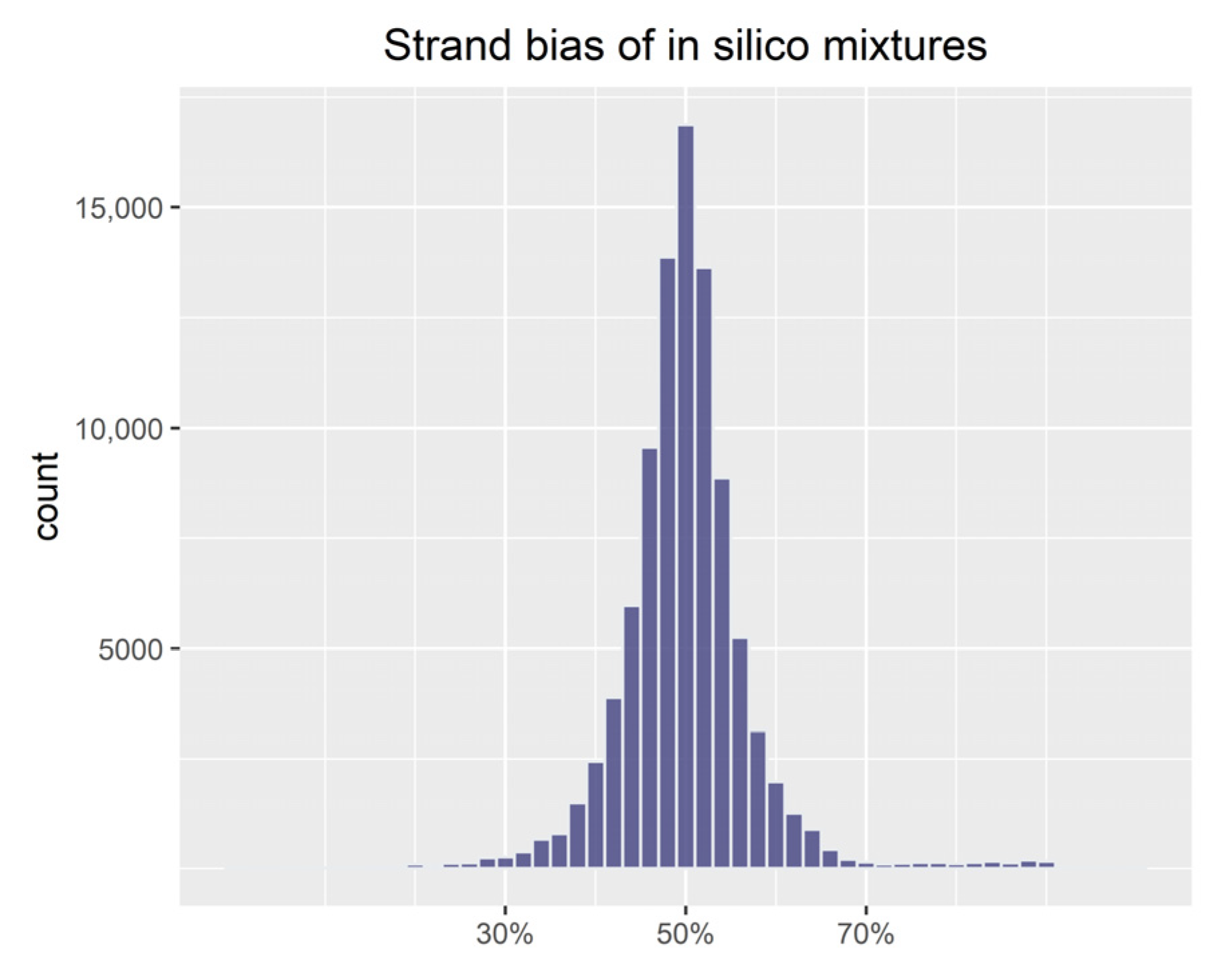
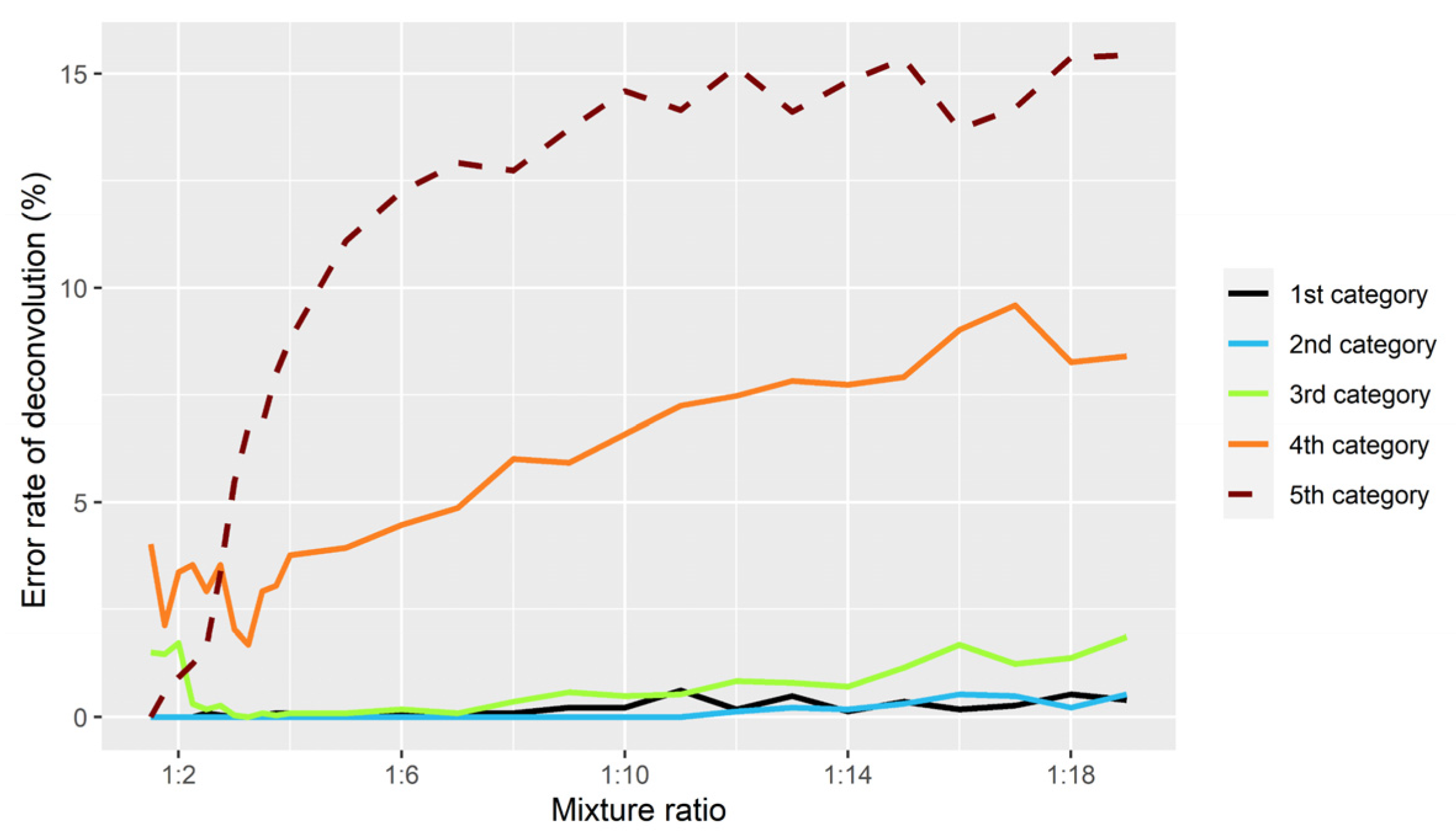
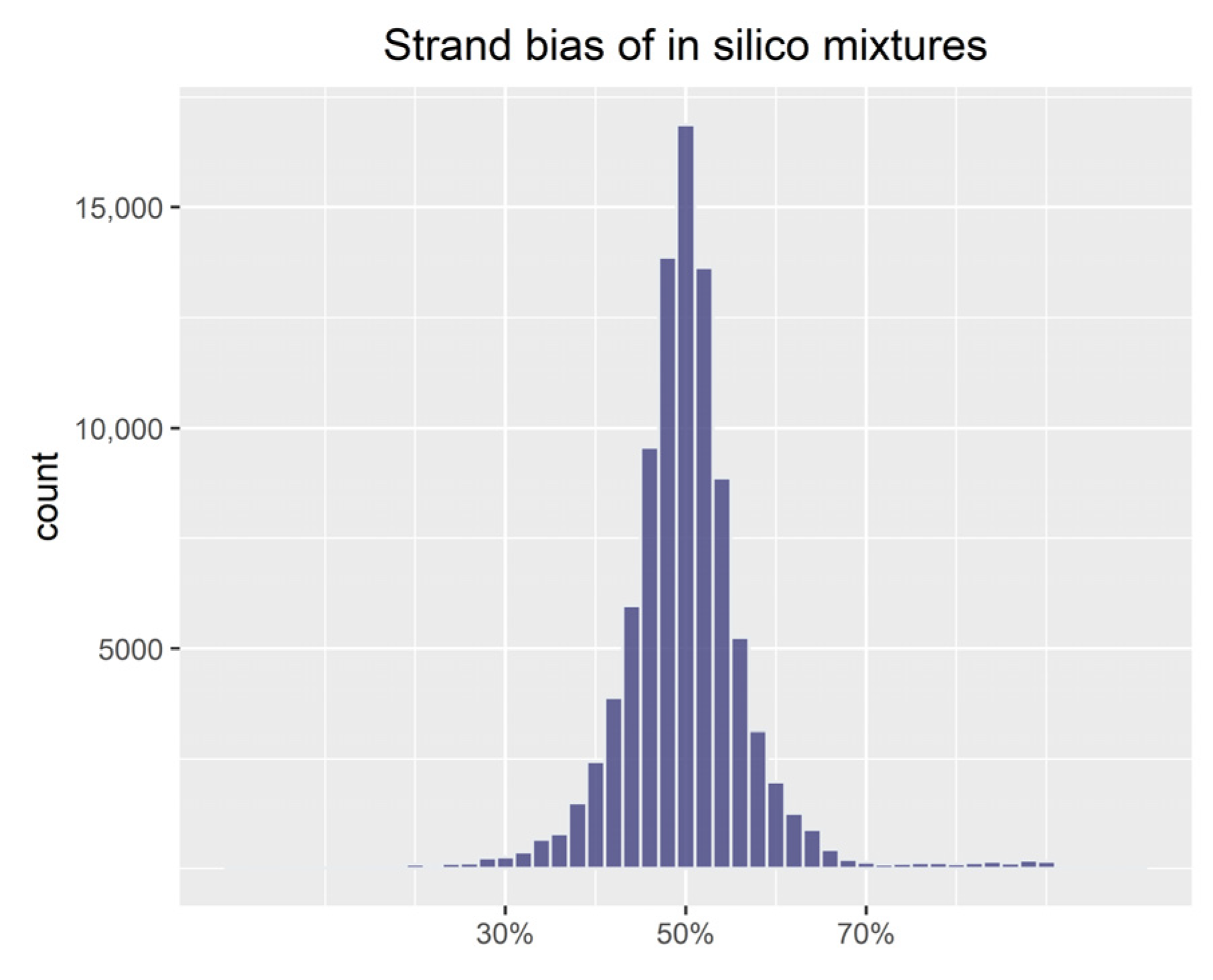
3.1. Evaluation of Simulations
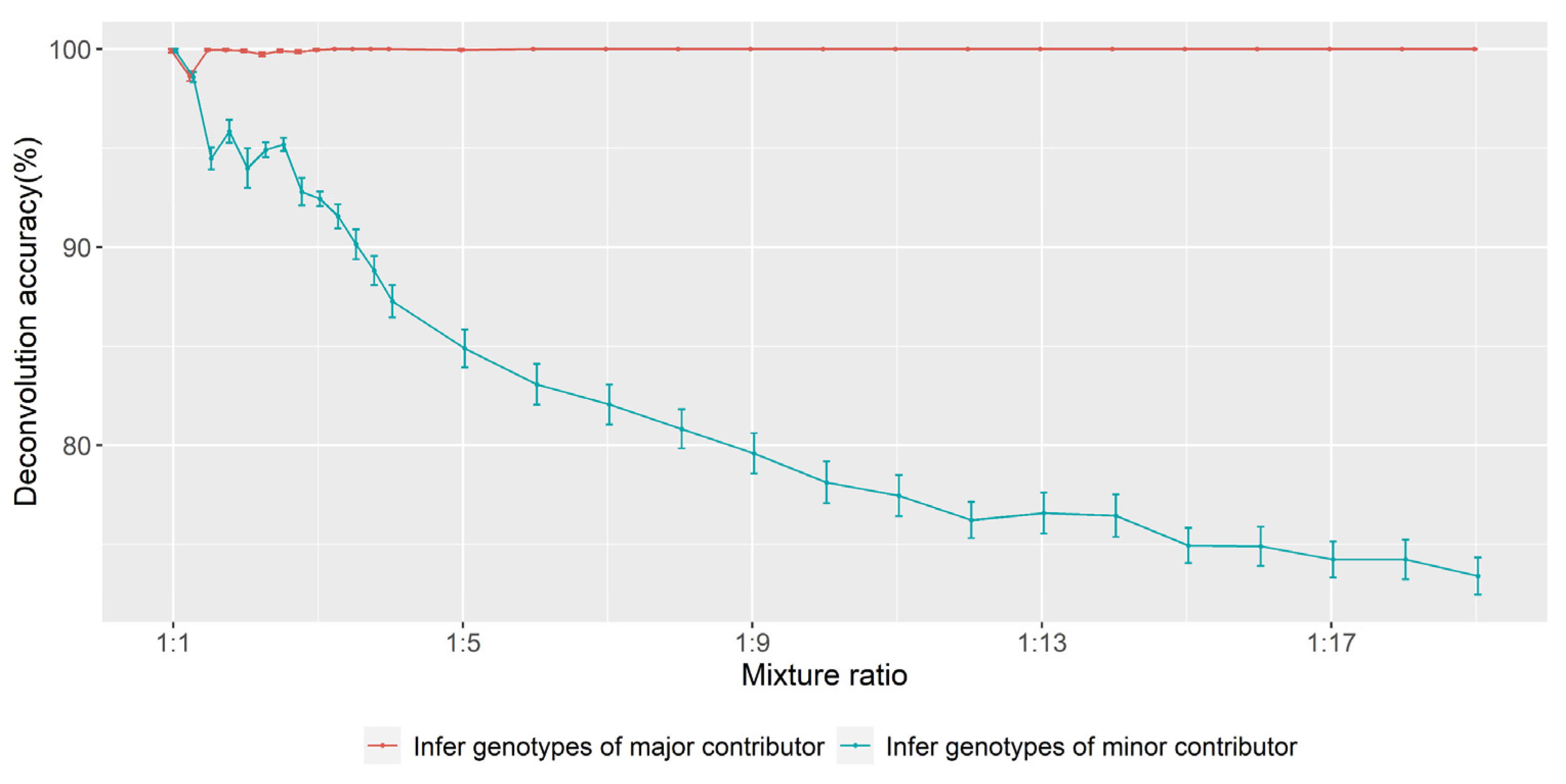
3.2. Accuracy of Inferring Genotypes
3.2.1. A-SNP
3.2.2. Y-SNP
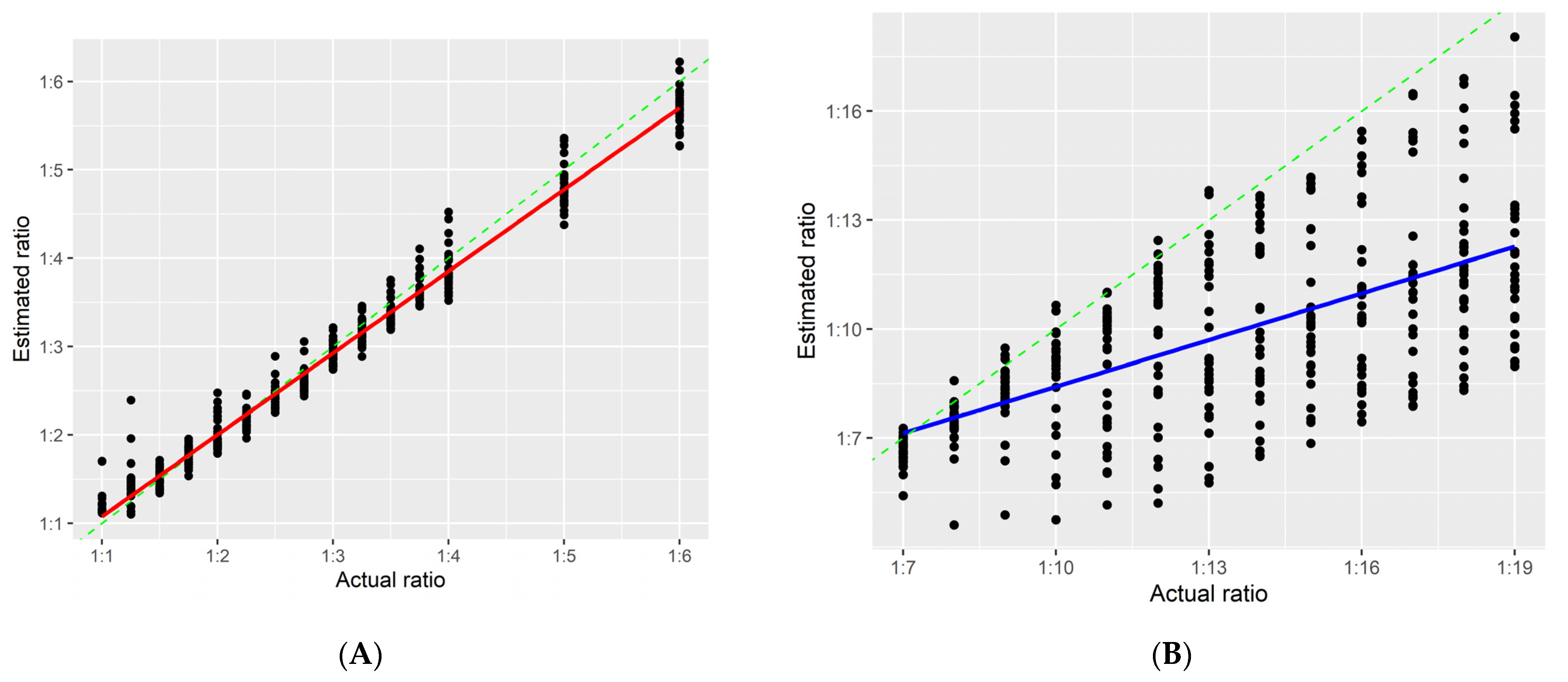
3.3. Estimated Ratio
3.4. Validation
3.5. Determination of Major and Minor Contributors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bieber, F.R.; Buckleton, J.S.; Budowle, B.; Butler, J.M.; Coble, M.D. Evaluation of forensic DNA mixture evidence: Protocol for evaluation, interpretation, and statistical calculations using the combined probability of inclusion. BMC Genet. 2016, 17, 125. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Xue, N.; Birdwell, J.D. Least-square deconvolution: A framework for interpreting short tandem repeat mixtures. J. Forensic Sci. 2006, 51, 1284–1297. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S.; Triggs, C.M.; Starling, L.; Stowell, L.I.; Walsh, K.A.J.; Buckleton, J. Interpreting DNA mixtures. J. Forensic Sci. 1997, 42, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Cowell, R.G.; Graversen, T.S.; Lauritzen, L.; Mortera, J. Analysis of forensic DNA mixtures with artefacts. J. Roy. Stat. Soc. Ser. C. Appl. Stat. 2015, 64, 1–48. [Google Scholar] [CrossRef]
- Oldoni, F.; Podini, D. Forensic molecular biomarkers for mixture analysis. Forensic Sci. Int. Genet. 2019, 41, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.; Haned, H.; Bleka, O.; Hansson, O.; Dorum, G.; Egeland, T. Genotyping and interpretation of STR-DNA: Low-template, mixtures and database matches-Twenty years of research and development. Forensic Sci. Int. Genet. 2015, 18, 100–117. [Google Scholar] [CrossRef]
- Gill, P.; Benschop, C.; Buckleton, J.; Bleka, O.; Taylor, D. A Review of Probabilistic Genotyping Systems: EuroForMix, DNAStatistX and STRmix. Genes 2021, 12, 1559. [Google Scholar] [CrossRef]
- Sharma, V.; Young, B.; Armogida, L.; Khan, A.; Wurmbach, E. Evaluation of ArmedXpert software tools, MixtureAce and Mixture Interpretation, to analyze MPS-STR data. Forensic Sci. Int. Genet. 2022, 56, 102603. [Google Scholar] [CrossRef]
- Clayton, T.M.; Whitaker, J.P.; Sparkes, R.; Gill, P. Analysis and interpretation of mixed forensic stains using DNA STR profiling. Forensic Sci. Int. 1998, 91, 55–70. [Google Scholar] [CrossRef]
- Pfeifer, C.M.; Klein-Unseld, R.; Klintschar, M.; Wiegand, P. Comparison of different interpretation strategies for low template DNA mixtures. Forensic Sci. Int. Genet. 2012, 6, 716–722. [Google Scholar] [CrossRef]
- Benschop, C.; Haned, H.; Sijen, T. Consensus and pool profiles to assist in the analysis and interpretation of complex low template DNA mixtures. Int. J. Legal Med. 2013, 127, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Boonyarit, H.; Mahasirimongkol, S.; Chavalvechakul, N.; Aoki, M.; Amitani, H.; Hosono, N.; Kamatani, N.; Kubo, M.; Lertrit, P. Development of a SNP set for human identification: A set with high powers of discrimination which yields high genetic information from naturally degraded DNA samples in the Thai population. Forensic Sci. Int. Genet. 2014, 11, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.M.; Coble, M.D.; Vallone, P.M. STRs vs. SNPs: Thoughts on the future of forensic DNA testing. Forensic Sci. Med. Pathol. 2007, 3, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Budowle, B.; van Daal, A. Forensically relevant SNP classes. Biotechniques 2008, 44, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Wang, S.; Zhang, J.; Zhang, J.; Yang, Z.; Sheng, X.; Hou, Y.; Zhang, S.; Li, C. Separation/extraction, detection, and interpretation of DNA mixtures in forensic science (review). Int. J. Legal Med. 2018, 132, 1247–1261. [Google Scholar] [CrossRef]
- Budowle, B.; Onorato, A.J.; Callaghan, T.F.; Della Manna, A.; Gross, A.M.; Guerrieri, R.A.; Luttman, J.C.; McClure, D.L. Mixture interpretation: Defining the relevant features for guidelines for the assessment of mixed DNA profiles in forensic casework. J. Forensic Sci. 2009, 54, 810–821. [Google Scholar] [CrossRef]
- Seo, S.B.; King, J.L.; Warshauer, D.H.; Davis, C.P.; Ge, J.; Budowle, B. Single nucleotide polymorphism typing with massively parallel sequencing for human identification. Int. J. Legal Med. 2013, 127, 1079–1086. [Google Scholar] [CrossRef]
- Borsting, C.; Morling, N. Next generation sequencing and its applications in forensic genetics. Forensic Sci. Int. Genet. 2015, 18, 78–89. [Google Scholar] [CrossRef]
- Gill, P. An assessment of the utility of single nucleotide polymorphisms (SNPs) for forensic purposes. Int. J. Legal Med. 2001, 114, 204–210. [Google Scholar] [CrossRef]
- Bleka, O.; Eduardoff, M.; Santos, C.; Phillips, C.; Parson, W.; Gill, P. Open source software EuroForMix can be used to analyse complex SNP mixtures. Forensic Sci. Int. Genet. 2017, 31, 105–110. [Google Scholar] [CrossRef]
- Hwa, H.L.; Wu, M.Y.; Chung, W.C.; Ko, T.M.; Lin, C.P.; Yin, H.I.; Lee, T.T.; Lee, J.C. Massively parallel sequencing analysis of nondegraded and degraded DNA mixtures using the ForenSeq system in combination with EuroForMix software. Int. J. Legal Med. 2019, 133, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.-W.; Li, Y.-H.; Chou, C.-F.; Lai, F.-P.; Chien, Y.-H.; Yin, H.-I.; Lee, T.-T.; Hwa, H.-L. DNA mixture interpretation using linear regression and neural networks on massively parallel sequencing data of single nucleotide polymorphisms. Aust. J. Forensic Sci. 2021, 54, 150–162. [Google Scholar] [CrossRef]
- Pascali, V.L. A novel computational strategy to predict the value of the evidence in the SNP-based forensic mixtures. PLoS ONE 2021, 16, e0247344. [Google Scholar] [CrossRef] [PubMed]
- Pakstis, A.J.; Speed, W.C.; Fang, R.; Hyland, F.C.; Furtado, M.R.; Kidd, J.R.; Kidd, K.K. SNPs for a universal individual identification panel. Hum. Genet. 2010, 127, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.J.; Phillips, C.; Borsting, C.; Balogh, K.; Bogus, M.; Fondevila, M.; Harrison, C.D.; Musgrave-Brown, E.; Salas, A.; Syndercombe-Court, D.; et al. A multiplex assay with 52 single nucleotide polymorphisms for human identification. Electrophoresis 2006, 27, 1713–1724. [Google Scholar] [CrossRef] [PubMed]
- Karafet, T.M.; Mendez, F.L.; Meilerman, M.B.; Underhill, P.A.; Zegura, S.L.; Hammer, M.F. New binary polymorphisms reshape and increase resolution of the human Y chromosomal haplogroup tree. Genome Res. 2008, 18, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Zhou, Y.; Song, H.; Zhao, J.; Shen, H.; Zhao, B.; Liu, F.; Jiang, X. Next generation sequencing of SNPs using the HID-Ion AmpliSeq Identity Panel on the Ion Torrent PGM platform. Forensic Sci. Int. Genet. 2016, 25, 73–84. [Google Scholar] [CrossRef]
- Buchard, A.; Kampmann, M.L.; Poulsen, L.; Borsting, C.; Morling, N. ISO 17025 validation of a next-generation sequencing assay for relationship testing. Electrophoresis 2016, 37, 2822–2831. [Google Scholar] [CrossRef]
- Garcia, O.; Soto, A.; Yurrebaso, I. Allele frequencies and other forensic parameters of the HID-Ion AmpliSeq Identity Panel markers in Basques using the Ion Torrent PGM platform. Forensic Sci. Int. Genet. 2017, 28, e8–e10. [Google Scholar] [CrossRef]
- Li, R.; Zhang, C.; Li, H.; Wu, R.; Li, H.; Tang, Z.; Zhen, C.; Ge, J.; Peng, D.; Wang, Y.; et al. SNP typing using the HID-Ion AmpliSeq™ Identity Panel in a southern Chinese population. Int. J. Legal Med. 2018, 132, 997–1006. [Google Scholar] [CrossRef]
- Avila, E.; Felkl, A.B.; Graebin, P.; Nunes, C.P.; Alho, C.S. Forensic characterization of Brazilian regional populations through massive parallel sequencing of 124 SNPs included in HID ion Ampliseq Identity Panel. Forensic Sci. Int. Genet. 2019, 40, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Thermo Fisher Scientific. HID SNP Genotyper Plugin User Guide v5.2.2; Thermo Fisher Scientific: Waltham, MA, USA, 2017. [Google Scholar]
- Zhang, S.; Bian, Y.; Zhang, Z.; Zheng, H.; Wang, Z.; Zha, L.; Cai, J.; Gao, Y.; Ji, C.; Hou, Y.; et al. Parallel analysis of 124 universal SNPs for human identification by targeted semiconductor sequencing. Sci. Rep. 2015, 5, 18683. [Google Scholar] [CrossRef] [PubMed]
- Thermo Fisher Scientific. Precision ID SNP Panels with the HID Ion S5™/HID Ion GeneStudio™ S5 System Application Guide; Thermo Fisher Scientific: Waltham, MA, USA, 2019. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Dash, H.R.; Avila, E.; Jena, S.R.; Kaitholia, K.; Agarwal, R.; Alho, C.S.; Srivastava, A.; Singh, A.K. Forensic characterization of 124 SNPs in the central Indian population using precision ID Identity Panel through next-generation sequencing. Int. J. Legal Med. 2021, 136, 465–473. [Google Scholar] [CrossRef]
- Eduardoff, M.; Santos, C.; de la Puente, M.; Gross, T.E.; Fondevila, M.; Strobl, C.; Sobrino, B.; Ballard, D.; Schneider, P.M.; Carracedo, A.; et al. Inter-laboratory evaluation of SNP-based forensic identification by massively parallel sequencing using the Ion PGM. Forensic Sci. Int. Genet. 2015, 17, 110–121. [Google Scholar] [CrossRef]
- Borsting, C.; Fordyce, S.L.; Olofsson, J.; Mogensen, H.S.; Morling, N. Evaluation of the Ion Torrent HID SNP 169-plex: A SNP typing assay developed for human identification by second generation sequencing. Forensic Sci. Int. Genet. 2014, 12, 144–154. [Google Scholar] [CrossRef]
- Samtools. Available online: http://www.htslib.org (accessed on 9 October 2021).
- GNU Operating System. Available online: https://www.gnu.org/software/coreutils/manual/ (accessed on 9 October 2021).
- Samtools Mpileup. Available online: http://www.htslib.org/doc/samtools-mpileup.html (accessed on 9 October 2021).
- Dorum, G.; Kaur, N.; Gysi, M. Pedigree-based relationship inference from complex DNA mixtures. Int. J. Legal Med. 2017, 131, 629–641. [Google Scholar] [CrossRef]
- Slooten, K. Distinguishing between donors and their relatives in complex DNA mixtures with binary models. Forensic Sci. Int. Genet. 2016, 21, 95–109. [Google Scholar] [CrossRef]
- Taylor, D.; Bright, J.A.; Buckleton, J. Considering relatives when assessing the evidential strength of mixed DNA profiles. Forensic Sci. Int. Genet. 2014, 13, 259–263. [Google Scholar] [CrossRef]
- Tiedge, T.M.; Nagachar, N.; Wendt, F.R.; Lakhtakia, A.; Roy, R. High-throughput DNA sequencing of environmentally insulted latent fingerprints after visualization with nanoscale columnar-thin-film technique. Sci. Justice 2021, 61, 505–515. [Google Scholar] [CrossRef]
- Meiklejohn, K.A.; Robertson, J.M. Evaluation of the Precision ID Identity Panel for the Ion TorrentTM PGMTM sequencer. Forensic Sci. Int. Genet. 2017, 31, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.J.; Misra, R.V.; Dallman, T.J.; Constantinidou, C.; Gharbia, S.E.; Wain, J.; Pallen, M.J. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 2012, 30, 434–439. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Individual 1 | Individual 2 | Mixture Profile | M1 | M2 | TF (%) | Mixture Deconvolution |
|---|---|---|---|---|---|---|
| AA | AA | AAAA | A | - | 100 | identical Hom (M1/M1) |
| AA | AG | AAAG | A | G | 75 | Hom & Het (M1/M1 & M1/M2) |
| AA | GG | AAGG | A or G | G or A | 50 | different Hom (M1/M1 & M2/M2) |
| AG | AA | AAAG | A | G | 75 | Het & Hom (M1/M2 & M1/M1) |
| AG | AG | AAGG | A or G | G or A | 50 | identical Het (M1/M2) |
| AG | GG | AGGG | G | A | 75 | Het & Hom (M1/M2 & M1/M1) |
| GG | AA | AAGG | A or G | G or A | 50 | different Hom (M1/M1 & M2/M2) |
| GG | AG | AGGG | G | A | 75 | Hom & Het (M1/M1 & M1/M2) |
| GG | GG | GGGG | G | - | 100 | identical Hom (M1/M1) |
| kco | TF | Minor & Major |
|---|---|---|
| n + 1 | TF1 | identical Hom |
| n | TF2 | Het & Hom |
| n − 1 | TF3 | different Hom |
| 1 | TF4 | Hom & Het |
| 0 | TF5 | identical Het |
| 2800 M and 9948 | Accuracy (%) | Estimated Ratio |
|---|---|---|
| 19:1 | 58.62 | 11.17:1 |
| 9:1 | 87.37 | 8.84:1 |
| 4:1 | 89.66 | 3.56:1 |
| 1:1 | 98.85 | 1:1.06 |
| 1:4 | 100 | 1:4.04 |
| 1:9 | 100 | 1:5.23 |
| 1:19 | 100 | 1:7.95 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, Y.; Zhang, P.; Xing, Y. A New Computational Deconvolution Algorithm for the Analysis of Forensic DNA Mixtures with SNP Markers. Genes 2022, 13, 884. https://doi.org/10.3390/genes13050884
Yin Y, Zhang P, Xing Y. A New Computational Deconvolution Algorithm for the Analysis of Forensic DNA Mixtures with SNP Markers. Genes. 2022; 13(5):884. https://doi.org/10.3390/genes13050884
Chicago/Turabian StyleYin, Yu, Peng Zhang, and Yu Xing. 2022. "A New Computational Deconvolution Algorithm for the Analysis of Forensic DNA Mixtures with SNP Markers" Genes 13, no. 5: 884. https://doi.org/10.3390/genes13050884
APA StyleYin, Y., Zhang, P., & Xing, Y. (2022). A New Computational Deconvolution Algorithm for the Analysis of Forensic DNA Mixtures with SNP Markers. Genes, 13(5), 884. https://doi.org/10.3390/genes13050884