
Polymerase ζ Is Involved in Mitochondrial DNA Maintenance Processes in Concert with APE1 Activity



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. siRNA Transfection
2.3. Treatments
2.4. Total Genomic DNA Isolation
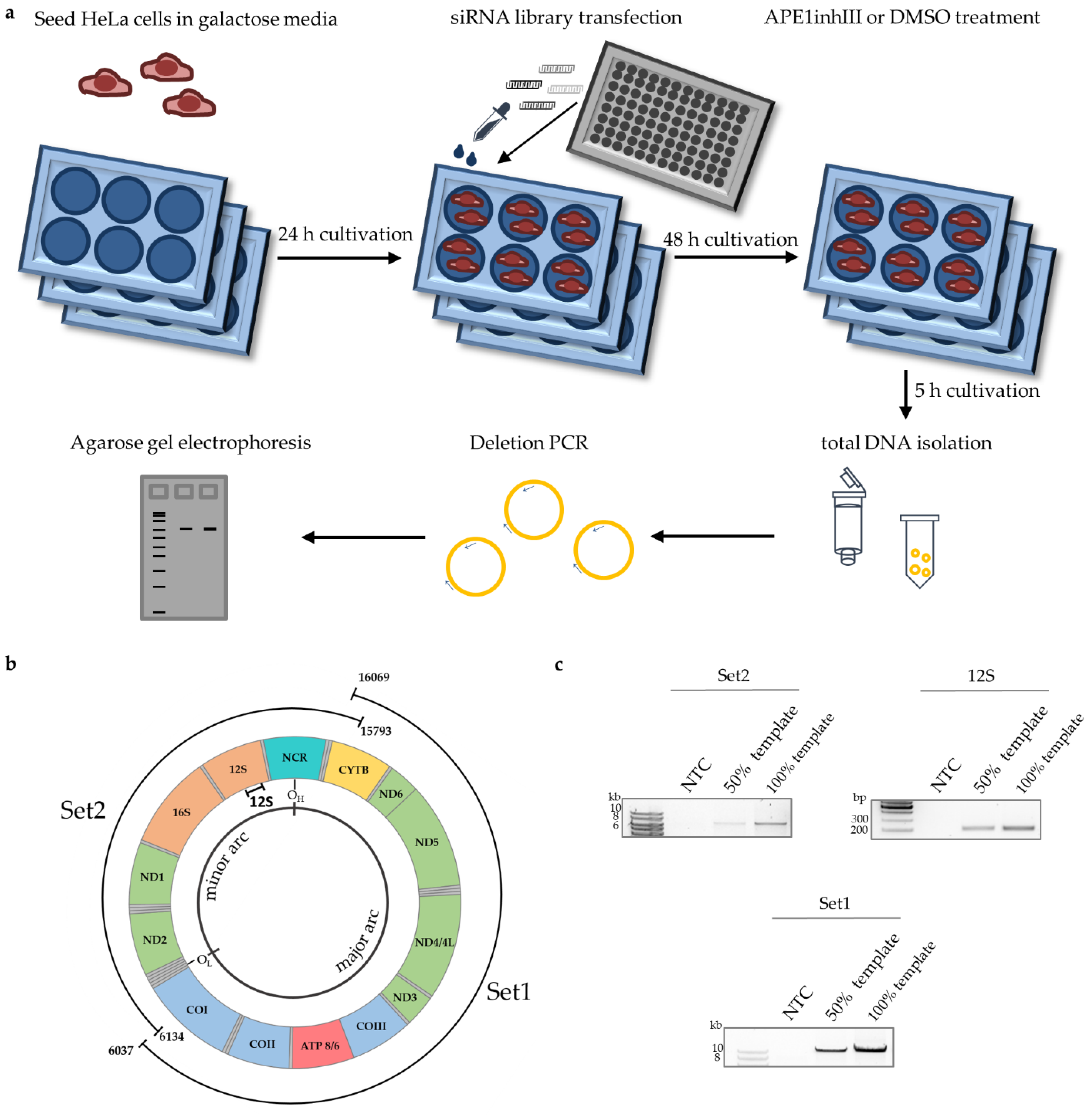
2.5. Long-Range PCR
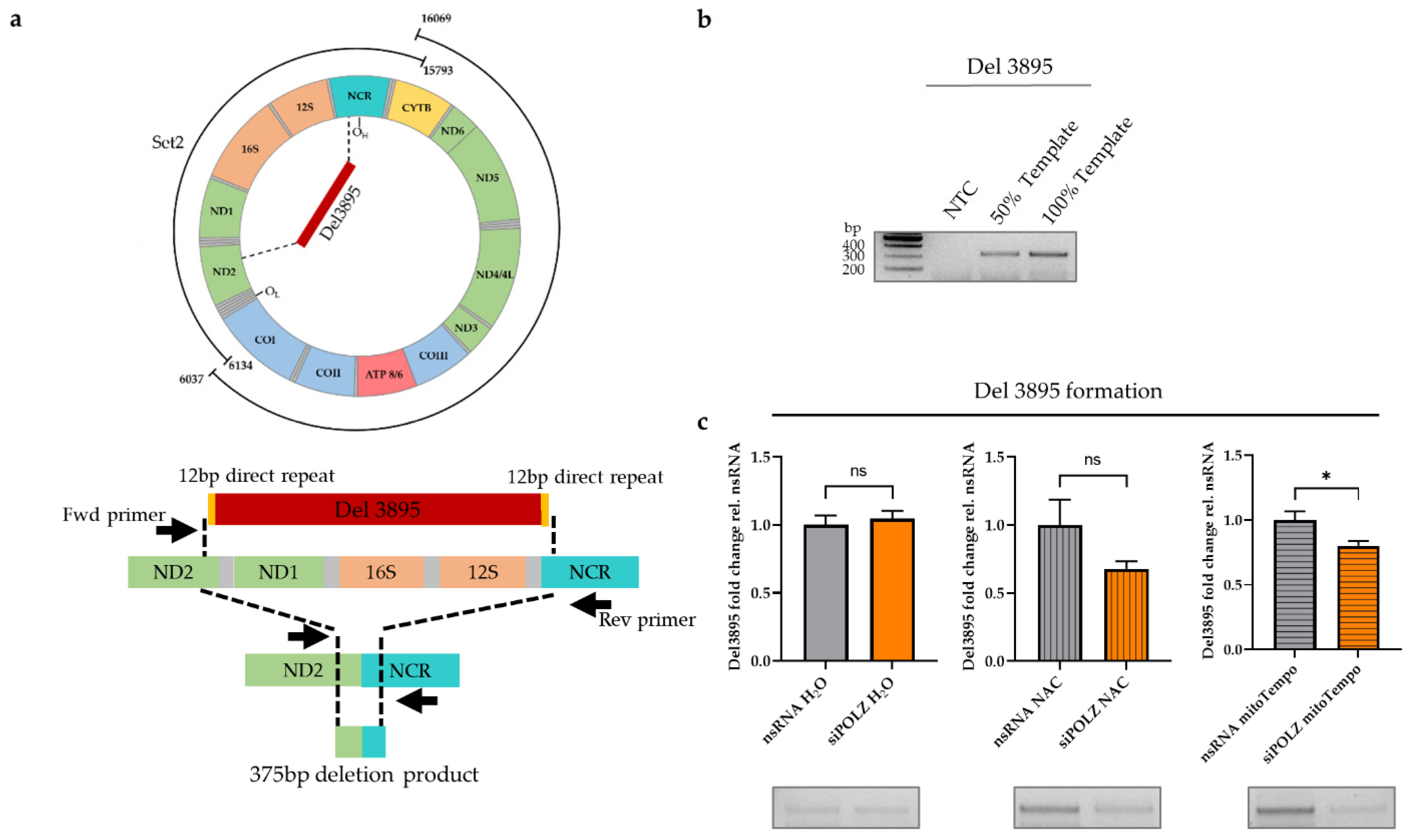
2.6. Del3895 PCR
2.7. Immunofluorescence Staining
2.8. MitoTracker Staining
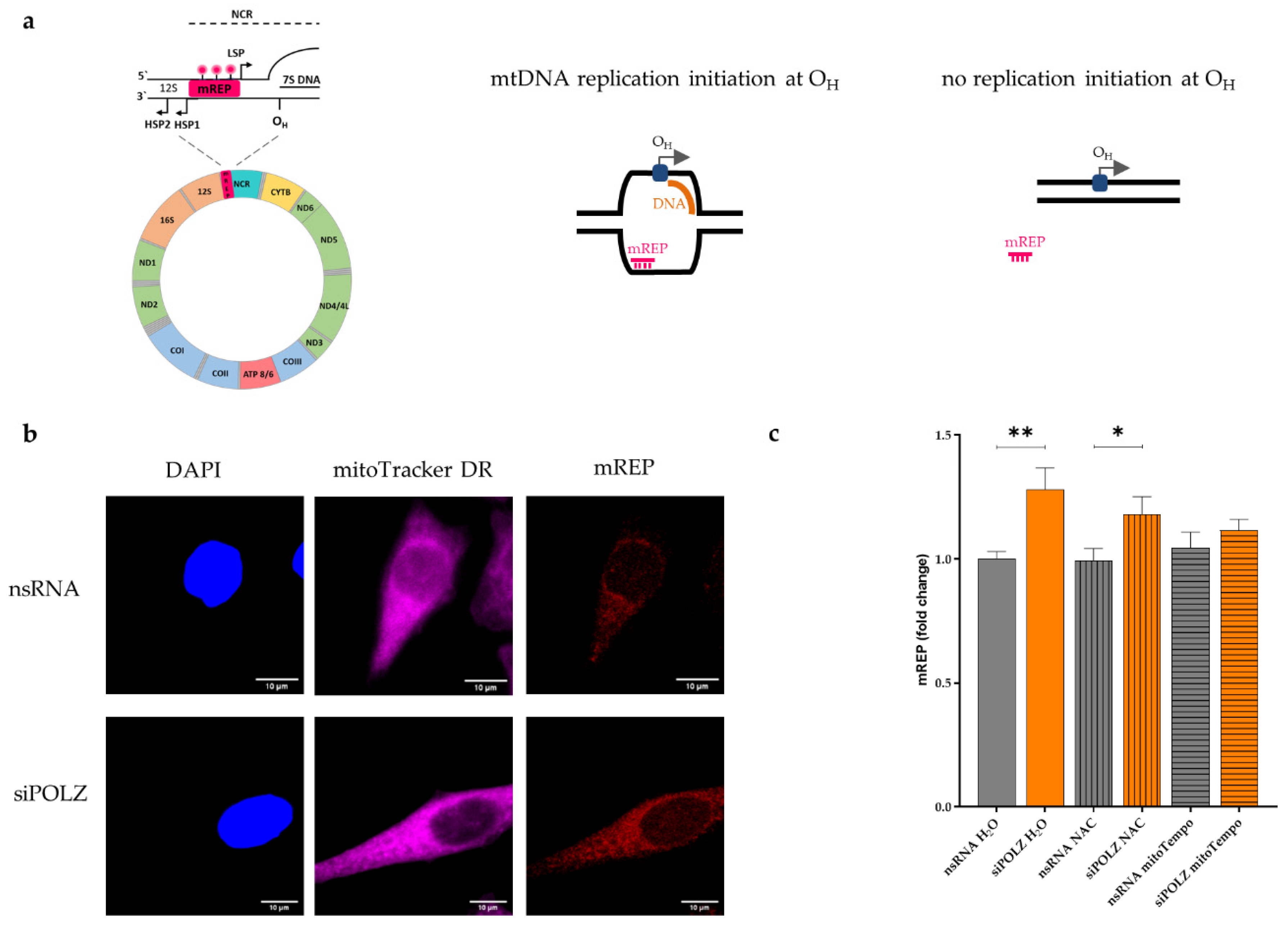
2.9. mTRIP Imaging Protocol
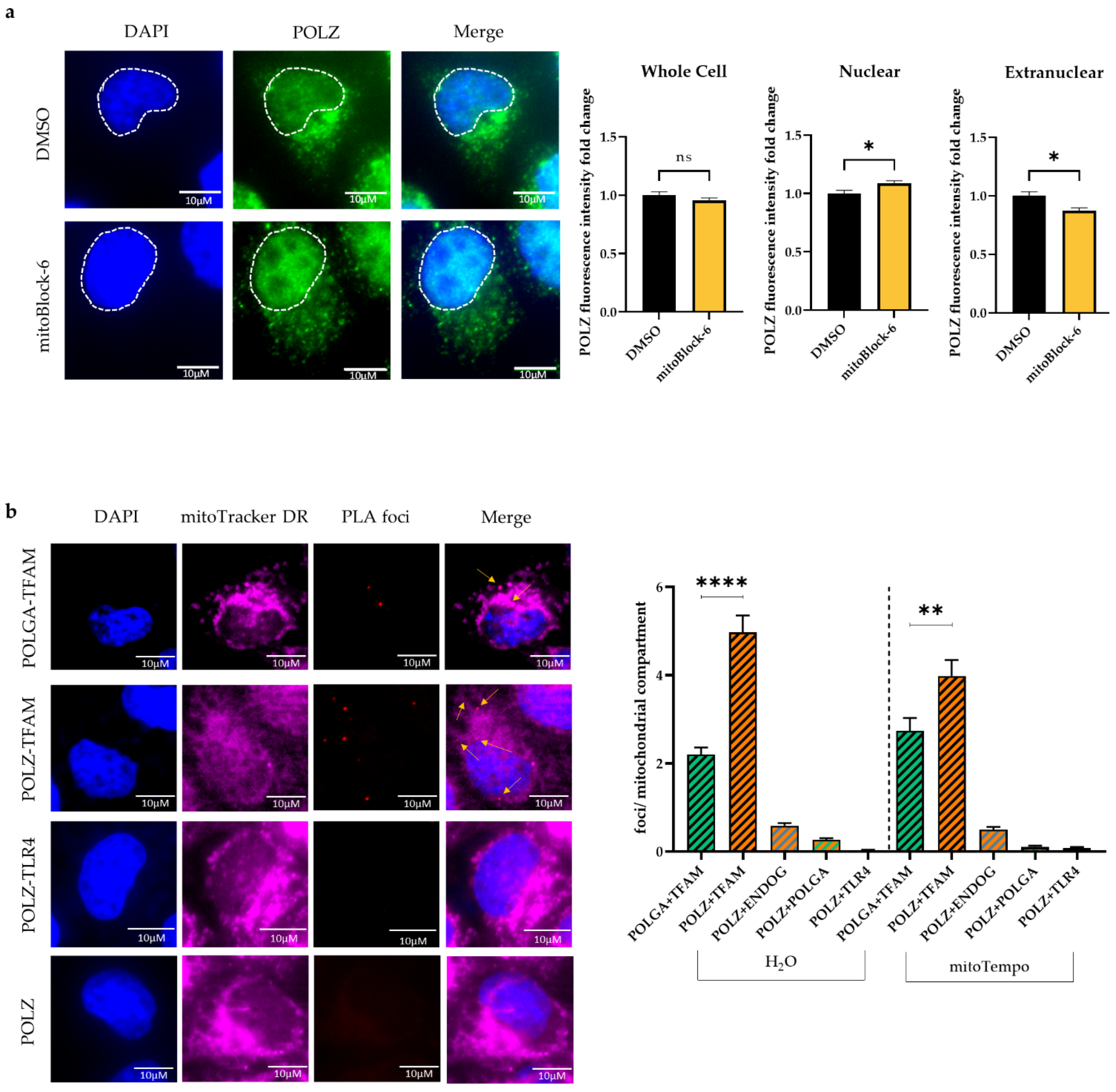
2.10. Proximity Ligation Assay (PLA)
2.11. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.12. Statistical Analysis
3. Results
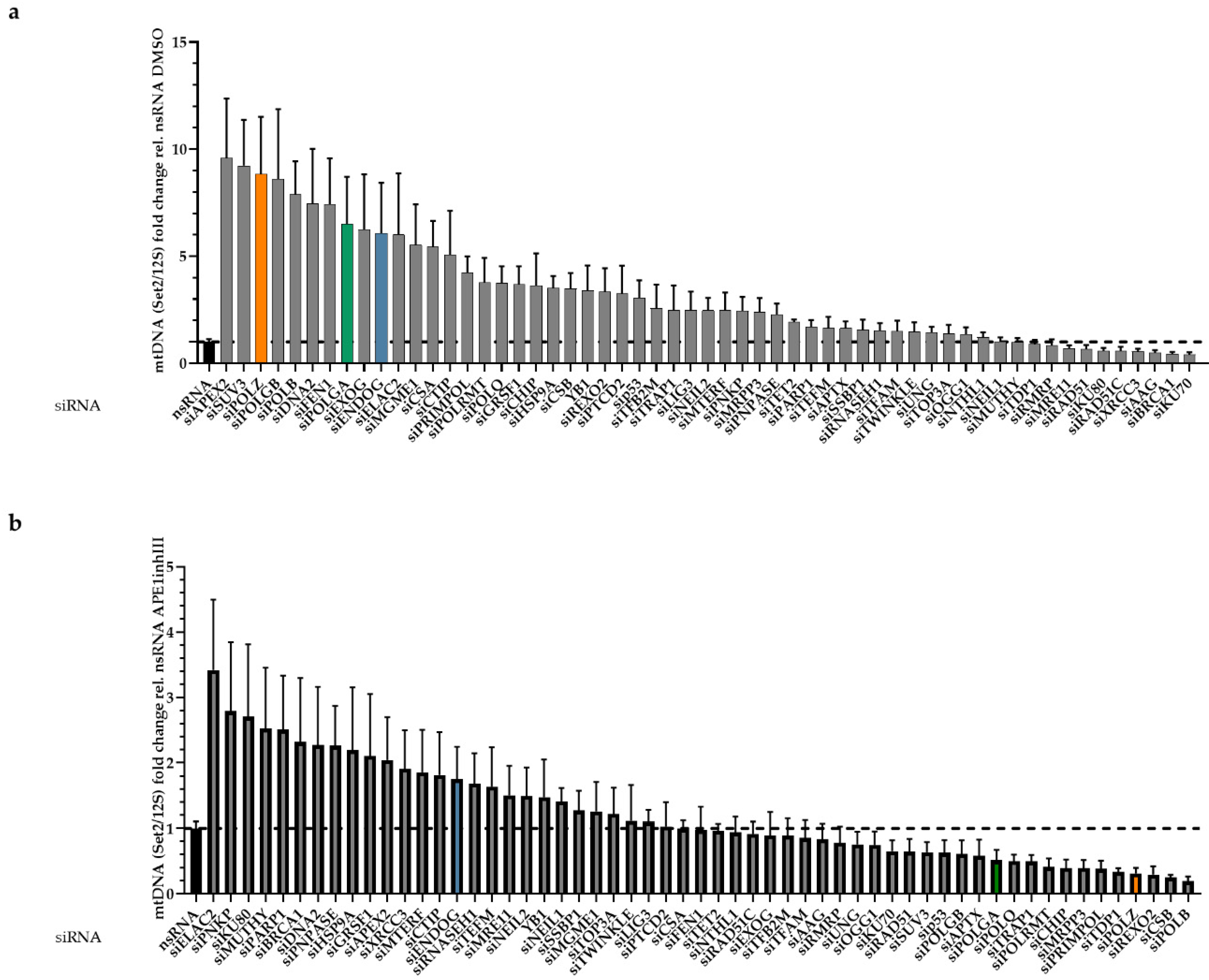
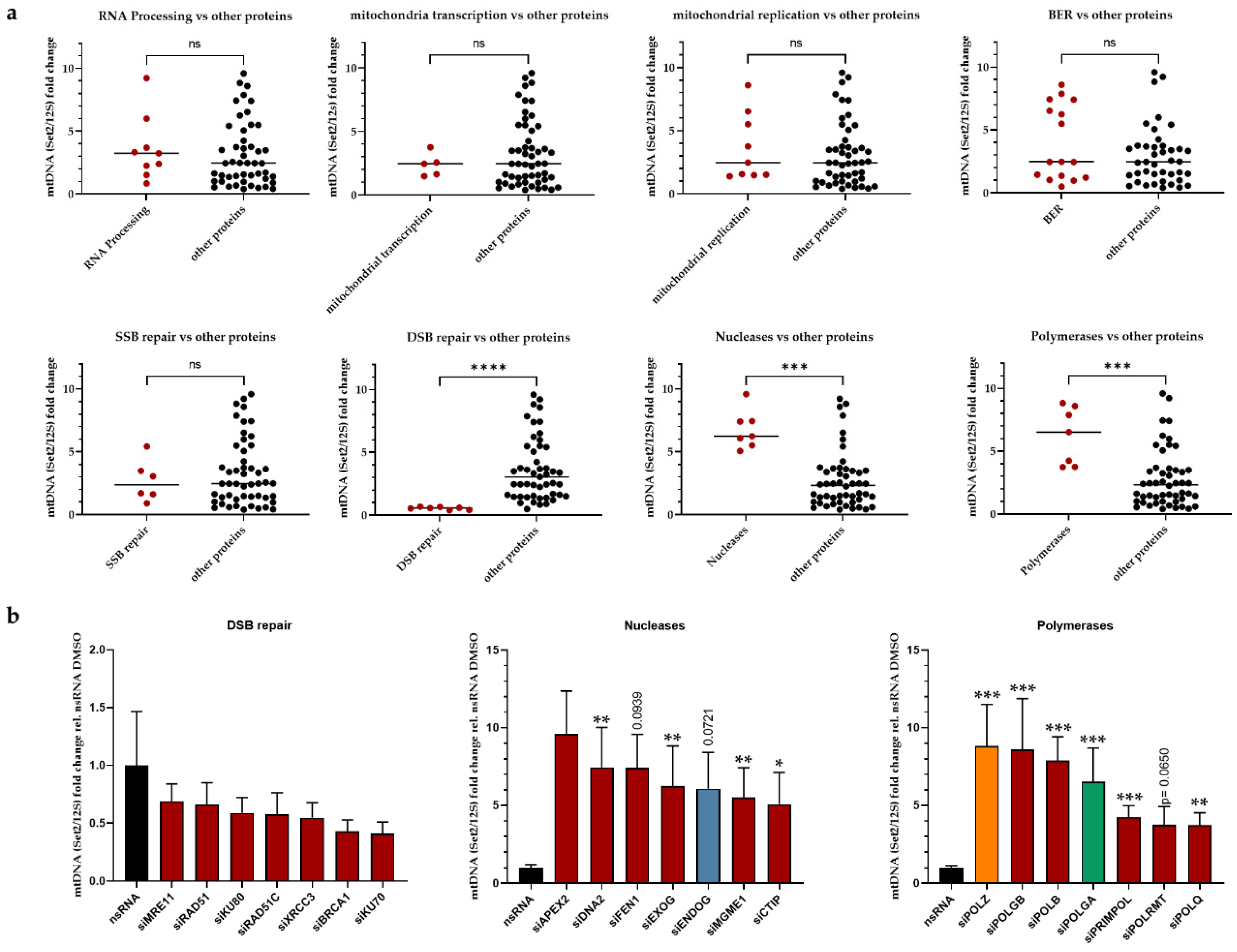
3.1. Identification of Proteins Involved in mtDNA Maintenaince
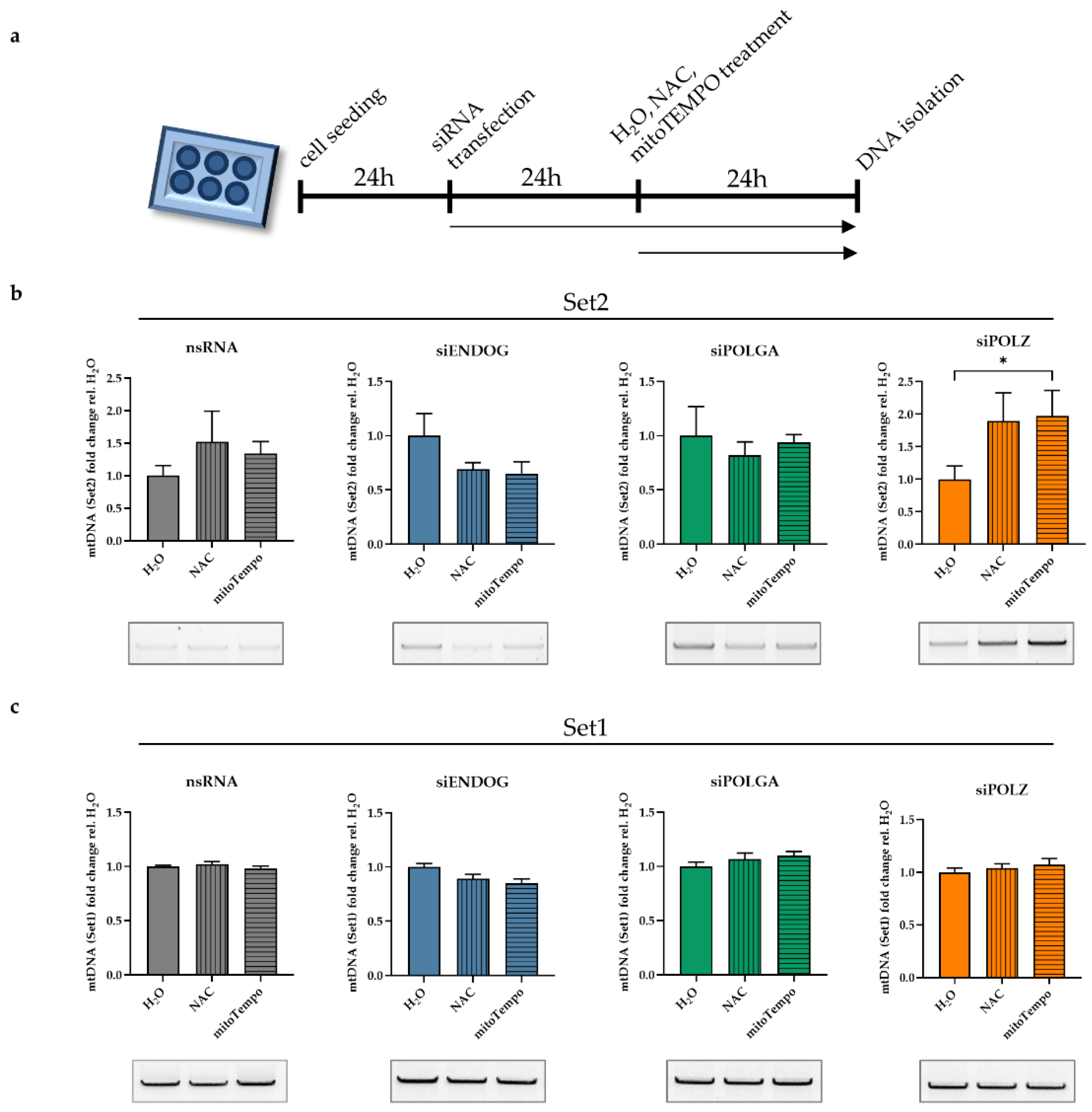
3.2. Roles of POLZ in mtDNA Stability as a Function of Mitochondrial ROS
3.3. POLZ Is Involved in 3895 bp Deletion Formation
3.4. POLZ Knockdown Stimulates mtDNA Replication Initiation
3.5. POLZ Localizes to Mitochondria
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Holt, I.J.; Speijer, D.; Kirkwood, T.B.L. The Road to Rack and Ruin: Selecting Deleterious Mitochondrial DNA Variants. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130451. [Google Scholar] [CrossRef] [PubMed]
- Fontana, G.A.; Gahlon, H.L. Mechanisms of Replication and Repair in Mitochondrial DNA Deletion Formation. Nucleic Acids Res. 2020, 48, 11244–11258. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, C.M.; Falkenberg, M.; Larsson, N.-G. Maintenance and Expression of Mammalian Mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, M.; Gustafsson, C.M. Mammalian Mitochondrial DNA Replication and Mechanisms of Deletion Formation. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 509–524. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Craigen, W.J.; Scaglia, F. Mitochondrial DNA Maintenance Defects. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1539–1555. [Google Scholar] [CrossRef]
- DeBalsi, K.L.; Hoff, K.E.; Copeland, W.C. Role of the Mitochondrial DNA Replication Machinery in Mitochondrial DNA Mutagenesis, Aging and Age-Related Diseases. Ageing Res. Rev. 2017, 33, 89–104. [Google Scholar] [CrossRef]
- Peter, B.; Falkenberg, M. TWINKLE and Other Human Mitochondrial DNA Helicases: Structure, Function and Disease. Genes 2020, 11, 408. [Google Scholar] [CrossRef]
- Kühl, I.; Miranda, M.; Posse, V.; Milenkovic, D.; Mourier, A.; Siira, S.J.; Bonekamp, N.A.; Neumann, U.; Filipovska, A.; Polosa, P.L.; et al. POLRMT Regulates the Switch between Replication Primer Formation and Gene Expression of Mammalian MtDNA. Sci. Adv. 2016, 2, e1600963. [Google Scholar] [CrossRef]
- Farr, C.L.; Wang, Y.; Kaguni, L.S. Functional Interactions of Mitochondrial DNA Polymerase and Single-Stranded DNA-Binding Protein. Template-Primer DNA Binding and Initiation and Elongation of DNA Strand Synthesis. J. Biol. Chem. 1999, 274, 14779–14785. [Google Scholar] [CrossRef]
- Korhonen, J.A.; Gaspari, M.; Falkenberg, M. TWINKLE Has 5′ -> 3′ DNA Helicase Activity and Is Specifically Stimulated by Mitochondrial Single-Stranded DNA-Binding Protein. J. Biol. Chem. 2003, 278, 48627–48632. [Google Scholar] [CrossRef]
- Jiang, M.; Xie, X.; Zhu, X.; Jiang, S.; Milenkovic, D.; Misic, J.; Shi, Y.; Tandukar, N.; Li, X.; Atanassov, I.; et al. The Mitochondrial Single-Stranded DNA Binding Protein Is Essential for Initiation of MtDNA Replication. Sci. Adv. 2021, 7, eabf8631. [Google Scholar] [CrossRef] [PubMed]
- Shoffner, J.M.; Lott, M.T.; Voljavec, A.S.; Soueidan, S.A.; Costigan, D.A.; Wallace, D.C. Spontaneous Kearns-Sayre/Chronic External Ophthalmoplegia plus Syndrome Associated with a Mitochondrial DNA Deletion: A Slip-Replication Model and Metabolic Therapy. Proc. Natl. Acad. Sci. USA 1989, 86, 7952–7956. [Google Scholar] [CrossRef] [PubMed]
- Persson, Ö.; Muthukumar, Y.; Basu, S.; Jenninger, L.; Uhler, J.P.; Berglund, A.-K.; McFarland, R.; Taylor, R.W.; Gustafsson, C.M.; Larsson, E.; et al. Copy-Choice Recombination during Mitochondrial L-Strand Synthesis Causes DNA Deletions. Nat. Commun. 2019, 10, 759. [Google Scholar] [CrossRef] [PubMed]
- Baptiste, B.A.; Baringer, S.L.; Kulikowicz, T.; Sommers, J.A.; Croteau, D.L.; Brosh, R.M.; Bohr, V.A. DNA Polymerase β Outperforms DNA Polymerase γ in Key Mitochondrial Base Excision Repair Activities. DNA Repair 2021, 99, 103050. [Google Scholar] [CrossRef]
- Kaufman, B.A.; van Houten, B. POLB: A New Role of DNA Polymerase Beta in Mitochondrial Base Excision Repair. DNA Repair 2017, 60, A1–A5. [Google Scholar] [CrossRef]
- Prakash, A.; Doublié, S. Base Excision Repair in the Mitochondria. J. Cell. Biochem. 2015, 116, 1490–1499. [Google Scholar] [CrossRef]
- Lu, C.Y.; Lee, H.C.; Fahn, H.J.; Wei, Y.H. Oxidative Damage Elicited by Imbalance of Free Radical Scavenging Enzymes Is Associated with Large-Scale MtDNA Deletions in Aging Human Skin. Mutat. Res. 1999, 423, 11–21. [Google Scholar] [CrossRef]
- Santos, R.X.; Correia, S.C.; Zhu, X.; Smith, M.A.; Moreira, P.I.; Castellani, R.J.; Nunomura, A.; Perry, G. Mitochondrial DNA Oxidative Damage and Repair in Aging and Alzheimer’s Disease. Antioxid. Redox Signal. 2013, 18, 2444–2457. [Google Scholar] [CrossRef]
- Moretton, A.; Morel, F.; Macao, B.; Lachaume, P.; Ishak, L.; Lefebvre, M.; Garreau-Balandier, I.; Vernet, P.; Falkenberg, M.; Farge, G. Selective Mitochondrial DNA Degradation Following Double-Strand Breaks. PLoS ONE 2017, 12, e0176795. [Google Scholar] [CrossRef]
- Peeva, V.; Blei, D.; Trombly, G.; Corsi, S.; Szukszto, M.J.; Rebelo-Guiomar, P.; Gammage, P.A.; Kudin, A.P.; Becker, C.; Altmüller, J.; et al. Linear Mitochondrial DNA Is Rapidly Degraded by Components of the Replication Machinery. Nat. Commun. 2018, 9, 1727. [Google Scholar] [CrossRef]
- Nissanka, N.; Bacman, S.R.; Plastini, M.J.; Moraes, C.T. The Mitochondrial DNA Polymerase Gamma Degrades Linear DNA Fragments Precluding the Formation of Deletions. Nat. Commun. 2018, 9, 2491. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.J.; Reeve, A.K.; Samuels, D.C.; Chinnery, P.F.; Blackwood, J.K.; Taylor, R.W.; Wanrooij, S.; Spelbrink, J.N.; Lightowlers, R.N.; Turnbull, D.M. What Causes Mitochondrial DNA Deletions in Human Cells? Nat. Genet. 2008, 40, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Nissanka, N.; Minczuk, M.; Moraes, C.T. Mechanisms of Mitochondrial DNA Deletion Formation. Trends Genet. 2019, 35, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative Stress Induces Degradation of Mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L. Mitochondrial DNA Degradation: A Quality Control Measure for Mitochondrial Genome Maintenance and Stress Response. Enzymes 2019, 45, 311–341. [Google Scholar] [CrossRef] [PubMed]
- Li, L.Y.; Luo, X.; Wang, X. Endonuclease G Is an Apoptotic DNase When Released from Mitochondria. Nature 2001, 412, 95–99. [Google Scholar] [CrossRef]
- Liu, X.; He, Y.; Li, F.; Huang, Q.; Kato, T.A.; Hall, R.P.; Li, C.-Y. Redefining the Roles of Apoptotic Factors in Carcinogenesis. Mol. Cell. Oncol. 2016, 3, e1054550. [Google Scholar] [CrossRef][Green Version]
- Gole, B.; Mian, E.; Rall, M.; Wiesmüller, L. Base Excision Repair Proteins Couple Activation-Induced Cytidine Deaminase and Endonuclease G during Replication Stress-Induced MLL Destabilization. Leukemia 2018, 32, 159–167. [Google Scholar] [CrossRef]
- Wiehe, R.S.; Gole, B.; Chatre, L.; Walther, P.; Calzia, E.; Ricchetti, M.; Wiesmüller, L. Endonuclease G Promotes Mitochondrial Genome Cleavage and Replication. Oncotarget 2018, 9, 18309–18326. [Google Scholar] [CrossRef]
- Ferezin, C.d.C.; Basei, F.L.; Melo-Hanchuk, T.D.; Oliveira, A.L.; Peres de Oliveira, A.; Mori, M.P.; Souza-Pinto, N.C.; Kobarg, J. NEK5 Interacts with LonP1 and Its Kinase Activity Is Essential for the Regulation of Mitochondrial Functions and MtDNA Maintenance. FEBS Open Bio 2021, 11, 546–563. [Google Scholar] [CrossRef]
- Wang, S.-F.; Huang, K.-H.; Tseng, W.-C.; Lo, J.-F.; Li, A.F.-Y.; Fang, W.-L.; Chen, C.-F.; Yeh, T.-S.; Chang, Y.-L.; Chou, Y.-C.; et al. DNAJA3/Tid1 Is Required for Mitochondrial DNA Maintenance and Regulates Migration and Invasion of Human Gastric Cancer Cells. Cancers 2020, 12, 3463. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; He, K.; Zhang, Y.; Hua, X.; Feng, M.; Zhao, Z.; Sun, Y.; Jiang, Y.; Xia, Q. XRCC2 Repairs Mitochondrial DNA Damage and Fuels Malignant Behavior in Hepatocellular Carcinoma. Cancer Lett. 2021, 512, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.K.; Wood, R.D. DNA Polymerase ζ in DNA Replication and Repair. Nucleic Acids Res. 2019, 47, 8348–8361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chatterjee, A.; Singh, K.K. Saccharomyces Cerevisiae Polymerase Zeta Functions in Mitochondria. Genetics 2006, 172, 2683–2688. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Baruffini, E.; Serafini, F.; Ferrero, I.; Lodi, T. Overexpression of DNA Polymerase Zeta Reduces the Mitochondrial Mutability Caused by Pathological Mutations in DNA Polymerase Gamma in Yeast. PLoS ONE 2012, 7, e34322. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Li, X.; Owens, K.M.; Vanniarajan, A.; Liang, P.; Singh, K.K. Human REV3 DNA Polymerase Zeta Localizes to Mitochondria and Protects the Mitochondrial Genome. PLoS ONE 2015, 10, e0140409. [Google Scholar] [CrossRef]
- Xiang, T.; Du, L.; Pham, P.; Zhu, B.; Jiang, S. Nelfinavir, an HIV Protease Inhibitor, Induces Apoptosis and Cell Cycle Arrest in Human Cervical Cancer Cells via the ROS-Dependent Mitochondrial Pathway. Cancer Lett. 2015, 364, 79–88. [Google Scholar] [CrossRef]
- Medraño-Fernandez, I.; Bestetti, S.; Bertolotti, M.; Bienert, G.P.; Bottino, C.; Laforenza, U.; Rubartelli, A.; Sitia, R. Stress Regulates Aquaporin-8 Permeability to Impact Cell Growth and Survival. Antioxid. Redox Signal. 2016, 24, 1031–1044. [Google Scholar] [CrossRef]
- Park, W.H. The Effect of MAPK Inhibitors and ROS Modulators on Cell Growth and Death of H₂O₂-Treated HeLa Cells. Mol. Med. Rep. 2013, 8, 557–564. [Google Scholar] [CrossRef]
- Han, Y.H.; Yang, Y.M.; Kim, S.Z.; Park, W.H. Attenuation of MG132-Induced HeLa Cell Death by N-Acetyl Cysteine via Reducing Reactive Oxygen Species and Preventing Glutathione Depletion. Anticancer Res. 2010, 30, 2107–2112. [Google Scholar]
- Kakuda, T.N. Pharmacology of Nucleoside and Nucleotide Reverse Transcriptase Inhibitor-Induced Mitochondrial Toxicity. Clin. Ther. 2000, 22, 685–708. [Google Scholar] [CrossRef]
- Rocheteau, P.; Chatre, L.; Briand, D.; Mebarki, M.; Jouvion, G.; Bardon, J.; Crochemore, C.; Serrani, P.; Lecci, P.P.; Latil, M.; et al. Sepsis Induces Long-Term Metabolic and Mitochondrial Muscle Stem Cell Dysfunction Amenable by Mesenchymal Stem Cell Therapy. Nat. Commun. 2015, 6, 10145. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.J.; Harbottle, A.; Birch-Machin, M.A. The Use of a 3895 Bp Mitochondrial DNA Deletion as a Marker for Sunlight Exposure in Human Skin. J. Investig. Dermatol. 2004, 123, 1020–1024. [Google Scholar] [CrossRef]
- Chatre, L.; Ricchetti, M. Prevalent Coordination of Mitochondrial DNA Transcription and Initiation of Replication with the Cell Cycle. Nucleic Acids Res. 2013, 41, 3068–3078. [Google Scholar] [CrossRef] [PubMed]
- Chatre, L.; Ricchetti, M. Large Heterogeneity of Mitochondrial DNA Transcription and Initiation of Replication Exposed by Single-Cell Imaging. J. Cell Sci. 2013, 126, 914–926. [Google Scholar] [CrossRef] [PubMed]
- Chatre, L.; Ricchetti, M. MTRIP: An Imaging Tool to Investigate Mitochondrial DNA Dynamics in Physiology and Disease at the Single-Cell Resolution. Methods Mol. Biol. 2015, 1264, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Sia, E.A. Mitochondrial DNA Repair and Damage Tolerance. Front. Biosci. 2017, 22, 4525. [Google Scholar] [CrossRef]
- Rong, Z.; Tu, P.; Xu, P.; Sun, Y.; Yu, F.; Tu, N.; Guo, L.; Yang, Y. The Mitochondrial Response to DNA Damage. Front. Cell Dev. Biol. 2021, 9, 1212. [Google Scholar] [CrossRef]
- Gohil, V.M.; Sheth, S.A.; Nilsson, R.; Wojtovich, A.P.; Lee, J.H.; Perocchi, F.; Chen, W.; Clish, C.B.; Ayata, C.; Brookes, P.S.; et al. Nutrient-Sensitized Screening for Drugs That Shift Energy Metabolism from Mitochondrial Respiration to Glycolysis. Nat. Biotechnol. 2010, 28, 249–255. [Google Scholar] [CrossRef]
- Orlicka-Płocka, M.; Gurda-Wozna, D.; Fedoruk-Wyszomirska, A.; Wyszko, E. Circumventing the Crabtree Effect: Forcing Oxidative Phosphorylation (OXPHOS) via Galactose Medium Increases Sensitivity of HepG2 Cells to the Purine Derivative Kinetin Riboside. Apoptosis 2020, 25, 835–852. [Google Scholar] [CrossRef]
- Graziewicz, M.A.; Longley, M.J.; Copeland, W.C. DNA Polymerase γ in Mitochondrial DNA Replication and Repair. Chem. Rev. 2006, 106, 383–405. [Google Scholar] [CrossRef] [PubMed]
- Makarova, A.V.; Burgers, P.M. Eukaryotic DNA Polymerase ζ. DNA Repair 2015, 29, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Krasich, R.; Copeland, W.C. DNA Polymerases in the Mitochondria: A Critical Review of the Evidence. Front. Biosci. 2017, 22, 692–709. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative Stress, Mitochondrial Damage and Neurodegenerative Diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Murphy, M.P.; Holmgren, A.; Larsson, N.-G.; Halliwell, B.; Chang, C.J.; Kalyanaraman, B.; Rhee, S.G.; Thornalley, P.J.; Partridge, L.; Gems, D.; et al. Unraveling the Biological Roles of Reactive Oxygen Species. Cell Metab. 2011, 13, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic Targeting of Mitochondrial Superoxide in Hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.J.; Ahamed, M.; Alhadlaq, H.; Alrokayan, S. Pt-Coated Au Nanoparticle Toxicity Is Preferentially Triggered Via Mitochondrial Nitric Oxide/Reactive Oxygen Species in Human Liver Cancer (HepG2) Cells. ACS Omega 2021, 6, 15431–15441. [Google Scholar] [CrossRef]
- Abdullah-Al-Shoeb, M.; Sasaki, K.; Kikutani, S.; Namba, N.; Ueno, K.; Kondo, Y.; Maeda, H.; Maruyama, T.; Irie, T.; Ishitsuka, Y. The Late-Stage Protective Effect of Mito-TEMPO against Acetaminophen-Induced Hepatotoxicity in Mouse and Three-Dimensional Cell Culture Models. Antioxidants 2020, 9, 965. [Google Scholar] [CrossRef]
- Harbottle, A.; Birch-Machin, M.A. Real-Time PCR Analysis of a 3895 Bp Mitochondrial DNA Deletion in Nonmelanoma Skin Cancer and Its Use as a Quantitative Marker for Sunlight Exposure in Human Skin. Br. J. Cancer 2006, 94, 1887–1893. [Google Scholar] [CrossRef]
- Gendron, S.P.; Bastien, N.; Mallet, J.D.; Rochette, P.J. The 3895-Bp Mitochondrial DNA Deletion in the Human Eye: A Potential Involvement in Corneal Ageing and Macular Degeneration. Mutagenesis 2013, 28, 197–204. [Google Scholar] [CrossRef][Green Version]
- Ræder, S.; Nepal, A.; Bjørås, K.; Seelinger, M.; Kolve, R.; Nedal, A.; Müller, R.; Otterlei, M. APIM-Mediated REV3L–PCNA Interaction Important for Error Free TLS Over UV-Induced DNA Lesions in Human Cells. Int. J. Mol. Sci. 2018, 20, 100. [Google Scholar] [CrossRef] [PubMed]
- Maya-Mendoza, A.; Moudry, P.; Merchut-Maya, J.M.; Lee, M.; Strauss, R.; Bartek, J. High Speed of Fork Progression Induces DNA Replication Stress and Genomic Instability. Nature 2018, 559, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.-J.C. Compensatory Amplification of MtDNA in a Patient with a Novel Deletion/Duplication and High Mutant Load. J. Med. Genet. 2003, 40, e125. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Kim, S.H.; Hamasaki, N. Mitochondrial Transcription Factor A (TFAM): Roles in Maintenance of MtDNA and Cellular Functions. Mitochondrion 2007, 7, 39–44. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Bailis, J.M. DNA Damage and Decisions: CtIP Coordinates DNA Repair and Cell Cycle Checkpoints. Trends Cell Biol. 2010, 20, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Tadi, S.K.; Sebastian, R.; Dahal, S.; Babu, R.K.; Choudhary, B.; Raghavan, S.C. Microhomology-Mediated End Joining Is the Principal Mediator of Double-Strand Break Repair during Mitochondrial DNA Lesions. Mol. Biol. Cell 2016, 27, 223–235. [Google Scholar] [CrossRef]
- Wang, H.; Xu, X. Microhomology-Mediated End Joining: New Players Join the Team. Cell Biosci. 2017, 7, 6. [Google Scholar] [CrossRef]
- Lin, W.; Sampathi, S.; Dai, H.; Liu, C.; Zhou, M.; Hu, J.; Huang, Q.; Campbell, J.; Shin-Ya, K.; Zheng, L.; et al. Mammalian DNA2 Helicase/Nuclease Cleaves G-Quadruplex DNA and Is Required for Telomere Integrity. EMBO J. 2013, 32, 1425–1439. [Google Scholar] [CrossRef]
- Zheng, L.; Meng, Y.; Campbell, J.L.; Shen, B. Multiple Roles of DNA2 Nuclease/Helicase in DNA Metabolism, Genome Stability and Human Diseases. Nucleic Acids Res. 2020, 48, 16–35. [Google Scholar] [CrossRef]
- Falabella, M.; Fernandez, R.J.; Johnson, F.B.; Kaufman, B.A. Potential Roles for G-Quadruplexes in Mitochondria. Curr. Med. Chem. 2019, 26, 2918–2932. [Google Scholar] [CrossRef]
- Dong, D.W.; Pereira, F.; Barrett, S.P.; Kolesar, J.E.; Cao, K.; Damas, J.; Yatsunyk, L.A.; Johnson, F.B.; Kaufman, B.A. Association of G-Quadruplex Forming Sequences with Human MtDNA Deletion Breakpoints. BMC Genom. 2014, 15, 677. [Google Scholar] [CrossRef] [PubMed]
- Duguay, B.A.; Smiley, J.R. Mitochondrial Nucleases ENDOG and EXOG Participate in Mitochondrial DNA Depletion Initiated by Herpes Simplex Virus 1 UL12.5. J. Virol. 2013, 87, 11787–11797. [Google Scholar] [CrossRef] [PubMed]
- Dzierzbicki, P.; Kaniak-Golik, A.; Malc, E.; Mieczkowski, P.; Ciesla, Z. The Generation of Oxidative Stress-Induced Rearrangements in Saccharomyces Cerevisiae MtDNA Is Dependent on the Nuc1 (EndoG/ExoG) Nuclease and Is Enhanced by Inactivation of the MRX Complex. Mutat. Res. Fundam. Mol. Mech. Mutagenes. 2012, 740, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.-W.; Anderson, D. Reactive Oxygen Species-Induced DNA Damage and Its Modification: A Chemical Investigation. Mutat. Res. Fundam. Mol. Mech. Mutagenes. 1997, 379, 201–210. [Google Scholar] [CrossRef]
- Silva-Pinheiro, P.; Pardo-Hernández, C.; Reyes, A.; Tilokani, L.; Mishra, A.; Cerutti, R.; Li, S.; Rozsivalova, D.-H.; Valenzuela, S.; Dogan, S.A.; et al. DNA Polymerase Gamma Mutations That Impair Holoenzyme Stability Cause Catalytic Subunit Depletion. Nucleic Acids Res. 2021, 49, 5230–5248. [Google Scholar] [CrossRef] [PubMed]
- Ropp, P.A.; Copeland, W.C. Cloning and Characterization of the Human Mitochondrial DNA Polymerase, DNA Polymerase γ. Genomics 1996, 36, 449–458. [Google Scholar] [CrossRef]
- Wang, Z.; Song, Y.; Li, S.; Kurian, S.; Xiang, R.; Chiba, T.; Wu, X. DNA Polymerase θ (POLQ) Is Important for Repair of DNA Double-Strand Breaks Caused by Fork Collapse. J. Biol. Chem. 2019, 294, 3909–3919. [Google Scholar] [CrossRef]
- Black, S.J.; Ozdemir, A.Y.; Kashkina, E.; Kent, T.; Rusanov, T.; Ristic, D.; Shin, Y.; Suma, A.; Hoang, T.; Chandramouly, G.; et al. Molecular Basis of Microhomology-Mediated End-Joining by Purified Full-Length Polθ. Nat. Commun. 2019, 10, 4423. [Google Scholar] [CrossRef]
- Ray, S.; Breuer, G.; DeVeaux, M.; Zelterman, D.; Bindra, R.; Sweasy, J.B. DNA Polymerase Beta Participates in DNA End-Joining. Nucleic Acids Res. 2018, 46, 242–255. [Google Scholar] [CrossRef]
- Sykora, P.; Kanno, S.; Akbari, M.; Kulikowicz, T.; Baptiste, B.A.; Leandro, G.S.; Lu, H.; Tian, J.; May, A.; Becker, K.A.; et al. DNA Polymerase Beta Participates in Mitochondrial DNA Repair. Mol. Cell. Biol. 2017, 37, e00237-17. [Google Scholar] [CrossRef]
- Estep, K.N.; Butler, T.J.; Ding, J.; Brosh, R.M. G4-Interacting DNA Helicases and Polymerases: Potential Therapeutic Targets. Curr. Med. Chem. 2019, 26, 2881–2897. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, E.D.; Longley, M.J.; Copeland, W.C. Polymerase γ Efficiently Replicates through Many Natural Template Barriers but Stalls at the HSP1 Quadruplex. J. Biol. Chem. 2020, 295, 17802–17815. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.; Kopylov, M.; Gomez-Llorente, Y.; Jain, R.; Johnson, R.E.; Prakash, L.; Prakash, S.; Ubarretxena-Belandia, I.; Aggarwal, A.K. Structure and Mechanism of B-Family DNA Polymerase ζ Specialized for Translesion DNA Synthesis. Nat. Struct. Mol. Biol. 2020, 27, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Pustovalova, Y.; Magalhães, M.T.Q.; D’Souza, S.; Rizzo, A.A.; Korza, G.; Walker, G.C.; Korzhnev, D.M. Interaction between the Rev1 C-Terminal Domain and the PolD3 Subunit of Polζ Suggests a Mechanism of Polymerase Exchange upon Rev1/Polζ-Dependent Translesion Synthesis. Biochemistry 2016, 55, 2043–2053. [Google Scholar] [CrossRef]
- Taglialatela, A.; Leuzzi, G.; Sannino, V.; Cuella-Martin, R.; Huang, J.-W.; Wu-Baer, F.; Baer, R.; Costanzo, V.; Ciccia, A. REV1-Polζ Maintains the Viability of Homologous Recombination-Deficient Cancer Cells through Mutagenic Repair of PRIMPOL-Dependent SsDNA Gaps. Mol. Cell 2021, 81, 4008–4025.e7. [Google Scholar] [CrossRef]
- García-Gómez, S.; Reyes, A.; Martínez-Jiménez, M.I.; Chocrón, E.S.; Mourón, S.; Terrados, G.; Powell, C.; Salido, E.; Méndez, J.; Holt, I.J.; et al. PrimPol, an Archaic Primase/Polymerase Operating in Human Cells. Mol. Cell 2013, 52, 541–553. [Google Scholar] [CrossRef]
- Torregrosa-Muñumer, R.; Forslund, J.M.E.; Goffart, S.; Pfeiffer, A.; Stojkovič, G.; Carvalho, G.; Al-Furoukh, N.; Blanco, L.; Wanrooij, S.; Pohjoismäki, J.L.O. PrimPol Is Required for Replication Reinitiation after MtDNA Damage. Proc. Natl. Acad. Sci. USA 2017, 114, 11398–11403. [Google Scholar] [CrossRef]
- Maréchal, A.; Brisson, N. Recombination and the Maintenance of Plant Organelle Genome Stability. New Phytol. 2010, 186, 299–317. [Google Scholar] [CrossRef]
- Leffak, M. Break-Induced Replication Links Microsatellite Expansion to Complex Genome Rearrangements. BioEssays 2017, 39, 1700025. [Google Scholar] [CrossRef]
- Sakofsky, C.J.; Ayyar, S.; Deem, A.K.; Chung, W.-H.; Ira, G.; Malkova, A. Translesion Polymerases Drive Microhomology-Mediated Break-Induced Replication Leading to Complex Chromosomal Rearrangements. Mol. Cell 2015, 60, 860–872. [Google Scholar] [CrossRef]
- Madhusudan, S.; Smart, F.; Shrimpton, P.; Parsons, J.L.; Gardiner, L.; Houlbrook, S.; Talbot, D.C.; Hammonds, T.; Freemont, P.A.; Sternberg, M.J.E.; et al. Isolation of a Small Molecule Inhibitor of DNA Base Excision Repair. Nucleic Acids Res. 2005, 33, 4711–4724. [Google Scholar] [CrossRef] [PubMed]
- Roychoudhury, S.; Pramanik, S.; Harris, H.L.; Tarpley, M.; Sarkar, A.; Spagnol, G.; Sorgen, P.L.; Chowdhury, D.; Band, V.; Klinkebiel, D.; et al. Endogenous Oxidized DNA Bases and APE1 Regulate the Formation of G-Quadruplex StructuRes. in the Genome. Proc. Natl. Acad. Sci. USA 2020, 117, 11409–11420. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.; Raguseo, F.; Nuccio, S.P.; Liano, D.; Di Antonio, M. DNA G-Quadruplex Structures: More than Simple Roadblocks to Transcription? Nucleic Acids Res. 2021, 49, 8419–8431. [Google Scholar] [CrossRef]
- Moor, N.A.; Vasil’eva, I.A.; Anarbaev, R.O.; Antson, A.A.; Lavrik, O.I. Quantitative Characterization of Protein–Protein Complexes Involved in Base Excision DNA Repair. Nucleic Acids Res. 2015, 43, 6009–6022. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, H.; Lindahl, T. DNA Base Excision Repair. In Encyclopedia of Biological Chemistry; Elsevier: Amsterdam, The Netherlands, 2004; pp. 603–608. [Google Scholar]
- Torregrosa-Muñumer, R.; Hangas, A.; Goffart, S.; Blei, D.; Zsurka, G.; Griffith, J.; Kunz, W.S.; Pohjoismäki, J.L.O. Replication Fork Rescue in Mammalian Mitochondria. Sci. Rep. 2019, 9, 8785. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schreier, H.K.; Wiehe, R.S.; Ricchetti, M.; Wiesmüller, L. Polymerase ζ Is Involved in Mitochondrial DNA Maintenance Processes in Concert with APE1 Activity. Genes 2022, 13, 879. https://doi.org/10.3390/genes13050879
Schreier HK, Wiehe RS, Ricchetti M, Wiesmüller L. Polymerase ζ Is Involved in Mitochondrial DNA Maintenance Processes in Concert with APE1 Activity. Genes. 2022; 13(5):879. https://doi.org/10.3390/genes13050879
Chicago/Turabian StyleSchreier, Heike Katrin, Rahel Stefanie Wiehe, Miria Ricchetti, and Lisa Wiesmüller. 2022. "Polymerase ζ Is Involved in Mitochondrial DNA Maintenance Processes in Concert with APE1 Activity" Genes 13, no. 5: 879. https://doi.org/10.3390/genes13050879
APA StyleSchreier, H. K., Wiehe, R. S., Ricchetti, M., & Wiesmüller, L. (2022). Polymerase ζ Is Involved in Mitochondrial DNA Maintenance Processes in Concert with APE1 Activity. Genes, 13(5), 879. https://doi.org/10.3390/genes13050879