
12q21 Interstitial Deletions: Seven New Syndromic Cases Detected by Array-CGH and Review of the Literature

 , , , , , ,
, , , , , ,  , ,
, , 
 add
Show full author list
add
Show full author list
Abstract
:
1. Introduction
2. Materials and Methods
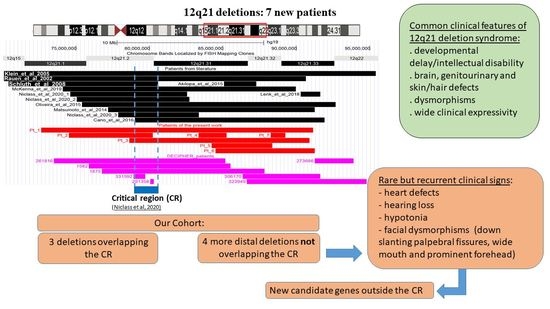
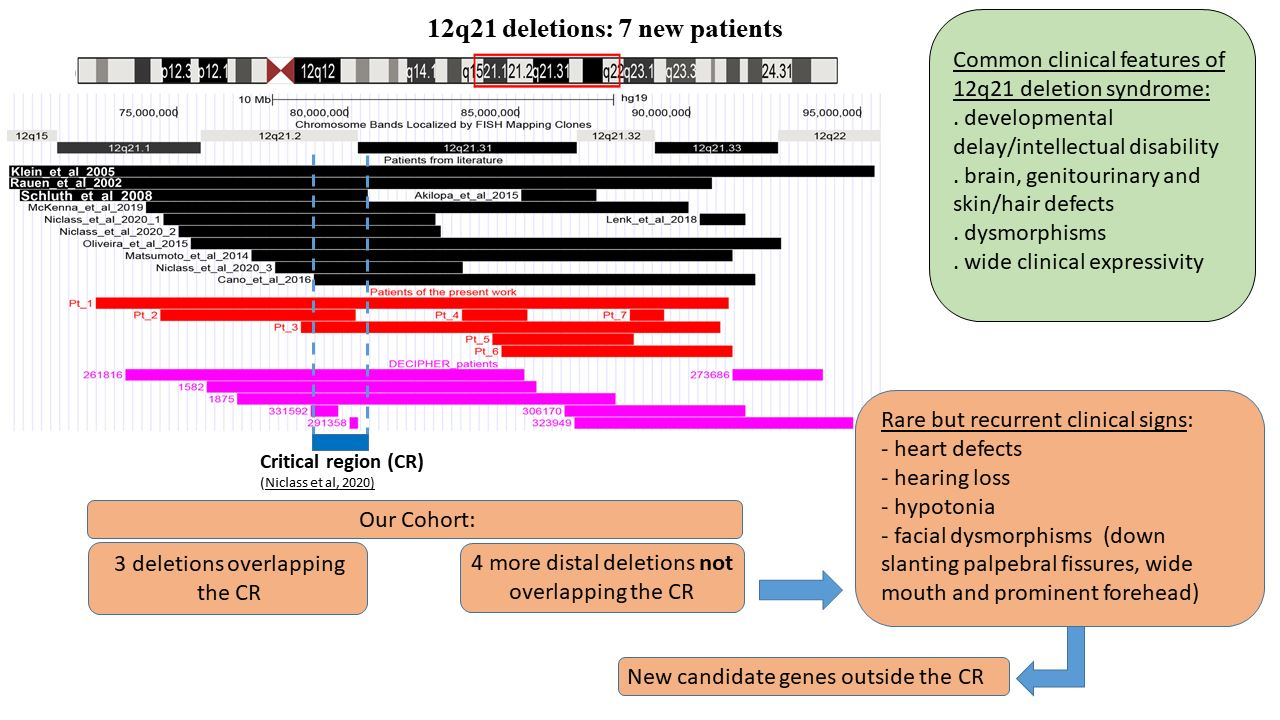
3. Results
3.1. Array-CGH
3.2. 12q21 Region and Synopsis of Relevant Genes
3.3. Literature and DECIPHER Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cano, M.; Trapasso, J.; Trapasso, T.; Matalon, R. 12q deletion with oculodentodigital dysplasia-like phenotype. Clin. Case Rep. Rev. 2016, 24, 387–390. [Google Scholar] [CrossRef] [Green Version]
- Klein, O.D.; Cotter, P.D.; Schmidt, A.M.; Bick, D.P.; Tidyman, W.E.; Albertson, D.G.; Pinkel, D.; Rauen, K.A. Interstitial deletion of chromosome 12q: Genotype-phenotype correlation of two patients utilizing array comparative genomic hybridization. Am. J. Med. Genet. A 2005, 138, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Mizuno, M.; Hamada, N.; Nozaki, Y.; Jimbo, E.F.; Momoi, M.Y.; Nagata, K.; Yamagata, T. LIN7A depletion disrupts cerebral cortex development, contributing to intellectual disability in 12q21-deletion syndrome. PLoS ONE 2014, 9, e92695. [Google Scholar] [CrossRef] [PubMed]
- McKenna, C.S.; Saxena, N.; Dabir, T.A.; Jones, J.; Smith, G.; Morrison, P.J. Phenotypic delineation of a 12q21 deletion syndrome. Clin. Dysmorphol. 2019, 28, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Niclass, T.; Le Guyader, G.; Beneteau, C.; Joubert, M.; Pizzuti, A.; Giuffrida, M.G.; Bernardini, L.; Gilbert-Dussardier, B.; Bilan, F.; Egloff, M. 12q21 deletion syndrome: Narrowing the critical region down to 1.6 Mb including SYT1 and PPP1R12A. Am. J. Med. Genet. A 2020, 182, 2133–2138. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, R.; Pereira, C.; Melo, J.B.; Mesquita, S.; Venâncio, M.; Carreira, I.M.; Saraiva, J. 12q21.2q22 deletion: A new patient. Am. J. Med. Genet. A 2015, 167A, 1877–1883. [Google Scholar] [CrossRef] [PubMed]
- Rauen, K.A.; Albertson, D.G.; Pinkel, D.; Cotter, P.D. Additional patient with del(12)(q21.2q22): Further evidence for a candidate region for cardio-facio-cutaneous syndrome? Am. J. Med. Genet. 2002, 110, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Schluth, C.; Gesny, R.; Borck, G.; Redon, R.; Abadie, V.; Kleinfinger, P.; Munnich, A.; Lyonnet, S.; Colleaux, L. New case of interstitial deletion 12(q15-q21.2) in a girl with facial dysmorphism and mental retardation. Am. J. Med. Genet. A 2008, 146A, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Akilapa, R.S.; Smith, K.; Balasubramanian, M. Clinical report: Inherited deletion of chromosome 12q21.31q21.32 associated with a distinct phenotype and intellectual disability. Clin. Dysmorphol. 2015, 24, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Lenk, J.; Porrmann, J.; Smitka, M.; Eger, I.; Schröck, E.; Hackmann, K.; Herber, R.; Raiskup, F.; Tzschach, A. Posterior amorphous corneal dystrophy in a patient with 12q21.33 deletion. Ophthalmic. Genet. 2018, 39, 645–647. [Google Scholar] [CrossRef] [PubMed]
- Lichter, P.; Cremer, T.; Borden, J.; Manuelidis, L.; Ward, D.C. Delineation of individual human chromosomes in metaphase and interphase cells by in situ suppression hybridization using recombinant DNA libraries. Hum. Genet. 1988, 80, 224–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UCSC Genome Browser. Available online: http://genome-euro.ucsc.edu/index.html (accessed on 15 January 2022).
- DECIPHER. Available online: https://www.deciphergenomics.org/ (accessed on 15 January 2022).
- OMIM. Available online: https://www.omim.org/ (accessed on 15 January 2022).
- gnomAD. Available online: https://gnomad.broadinstitute.org/ (accessed on 15 January 2022).
- Catusi, I.; Recalcati, M.P.; Bestetti, I.; Garzo, M.; Valtorta, C.; Alfonsi, M.; Alghisi, A.; Cappellani, S.; Casalone, R.; Caselli, R.; et al. Testing single/combined clinical categories on 5110 Italian patients with developmental phenotypes to improve array-based detection rate. Mol. Genet. Genomic. Med. 2020, 8, e1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Database of the Genomic Variants. Available online: http://dgv.tcag.ca/dgv/app/home (accessed on 15 January 2022).
- Baker, K.; Gordon, S.L.; Melland, H.; Bumbak, F.; Scott, D.J.; Jiang, T.J.; Owen, D.; Turner, B.J.; Boyd, S.G.; Rossi, M.; et al. SYT1-associated neurodevelopmental disorder: A case series. Brain 2018, 141, 2576–2591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, J.J.; Alkhunaizi, E.; Kruszka, P.; Pyle, L.C.; Grange, D.K.; Berger, S.I.; Payne, K.K.; Masser-Frye, D.; Hu, T.; Christie, M.R.; et al. Loss-of-Function Variants in PPP1R12A: From Isolated Sex Reversal to Holoprosencephaly Spectrum and Urogenital Malformations. Am. J. Hum. Genet. 2020, 106, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Uz, E.; Alanay, Y.; Aktas, D.; Vargel, I.; Gucer, S.; Tuncbilek, G.; von Eggeling, F.; Yilmaz, E.; Deren, O.; Posorski, N.; et al. Disruption of ALX1 causes extreme microphthalmia and severe facial clefting: Expanding the spectrum of autosomal-recessive ALX-related frontonasal dysplasia. Am. J. Hum. Genet. 2010, 86, 789–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sass, J.O.; Vaithilingam, J.; Gemperle-Britschgi, C.; Delnooz, C.C.; Kluijtmans, L.A.; van de Warrenburg, B.P.; Wevers, R.A. Expanding the phenotype in aminoacylase 1 (ACY1) deficiency: Characterization of the molecular defect in a 63-year-old woman with generalized dystonia. Metab. Brain Dis. 2016, 31, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Zazo Seco, C.; Serrão de Castro, L.; van Nierop, J.W.; Morín, M.; Jhangiani, S.; Verver, E.J.; Schraders, M.; Maiwald, N.; Wesdorp, M.; Venselaar, H.; et al. Allelic Mutations of KITLG, Encoding KIT Ligand, Cause Asymmetric and Unilateral Hearing Loss and Waardenburg Syndrome Type 2. Am. J. Hum. Genet. 2015, 97, 647–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]

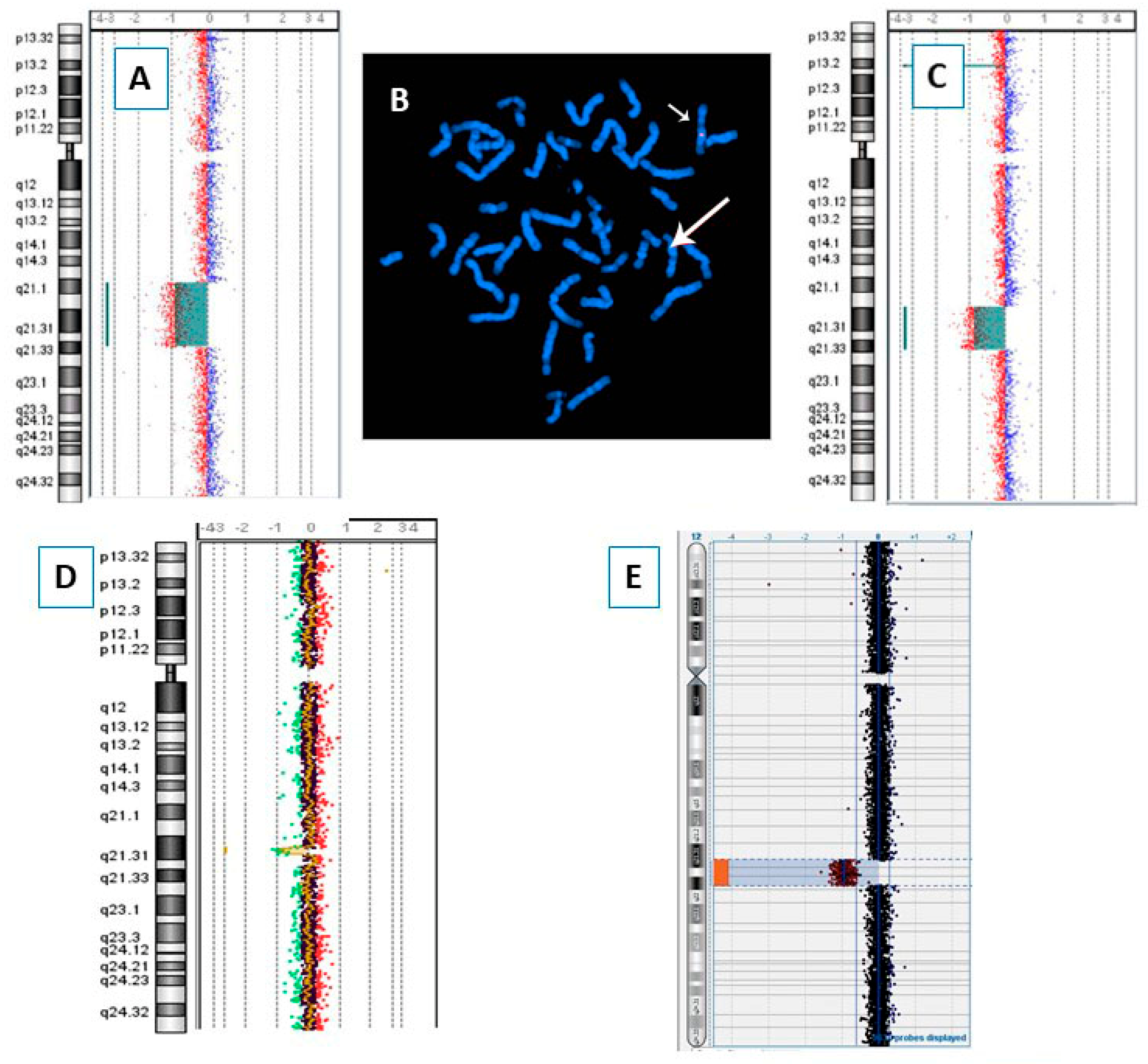

{kind=link}
{kind=link}
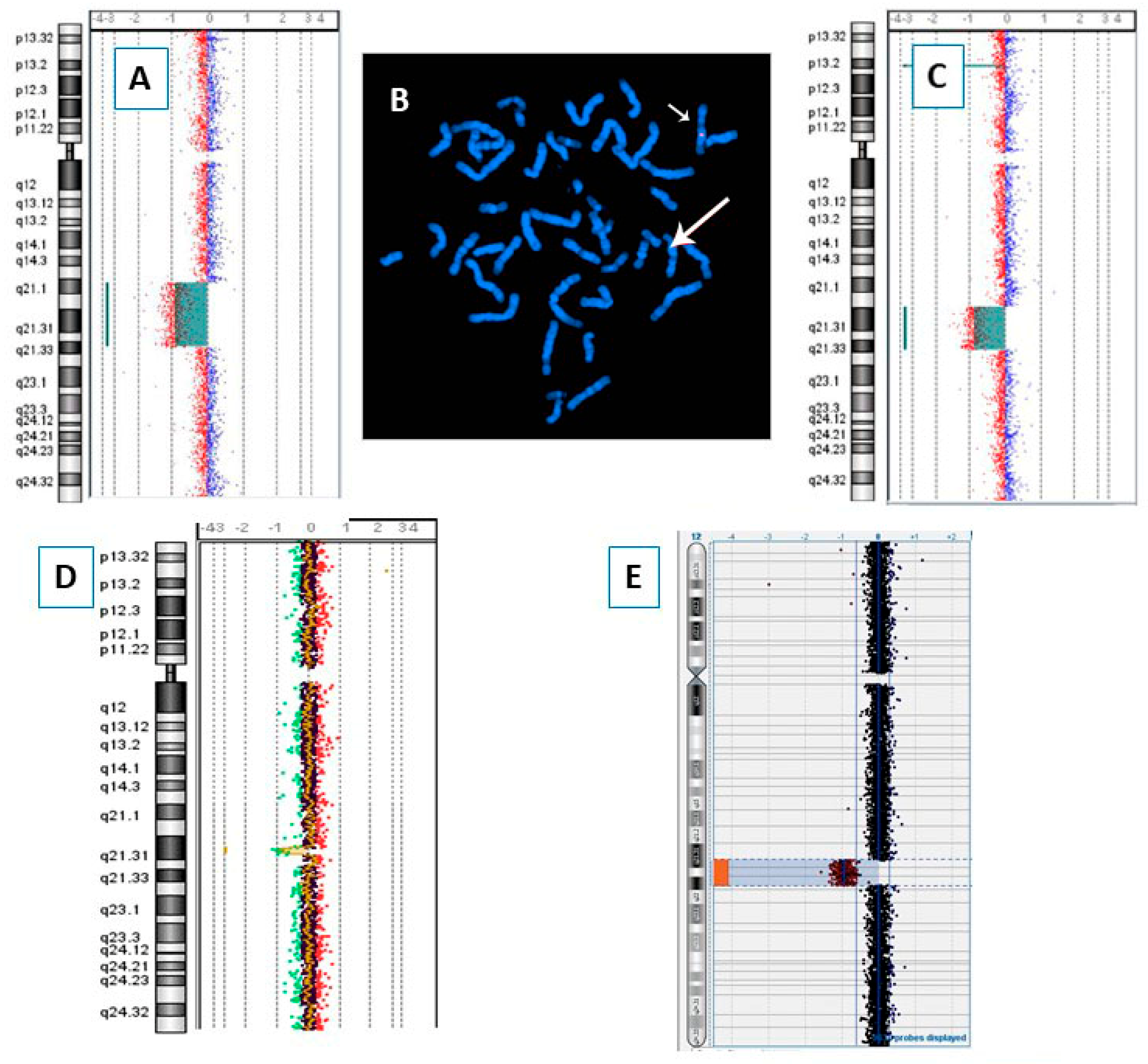{kind=link}
| Patients | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| Age at last evaluation and sex | 8y (M) | 11y (F) | 16y (M) | 11y (M) | 4y (M) | 7y (F) | 15y (M) |
| Coordinates on chromosome 12 (hg19) | 72634701-91163349 | 74536114-80234335 | 78634010-90918721 | 83359339-85260949 | 84234089-88380156 | 84507288-91264704 | 88264952-89267923 |
| Deletion size (Mb) | 18.53 | 5.7 | 12.28 | 1.9 | 4.15 | 6.76 | 1 |
| Inheritance | Unknown | From mother with similar phenotype | Unknown | Unknown | de novo | de novo | de novo |
| Growth retardation | + | - | - | - | + | - | - |
| DD/ID | Learning and language impairment. | Mild ID, language impairment. | Mild DD | DD | Mild ID. Receptive and expressive language significant delayed. | Learning and language impairments. | Mild ID and language impairment. |
| Prominent forehead | - | - | - | - | + | + | - |
| Hypertelorism | - | - | + | - | - | + | - |
| Low set ears | - | - | - | - | + | - | |
| Short nose | - | + | + | - | - | - | - |
| Other dysmorphisms | Relative macrocephaly. | Dysplastic ears, thick philtrum, large mouth. | - | - | Broad nasal base, low-hanging columella, wide mouth, down-slanting palpebral fissures. Relative microcephaly. | Upturned nose, long philtrum, ogival palate, wide mouth. | Ogival palate, eye asymmetry. |
| 2–3 toe syndactyly + single palmar crease | - | - | - | - | - | - | - |
| Cardiac anomalies | - | - | - | Ostium secundum atrial septal defect. | - | Small oval fossa shunt, interatrial shunt. | - |
| Ectodermal abnormalities | Dry skin, sparse eyebrows. | - | + | - | - | - | - |
| Ocular abnormalities | Astigmatism | Exophthalmos | + | - | Epichantus | Exophoria | Astigmatism |
| Genitourinary anomalies | Horseshoe kidney | - | - | - | - | - | - |
| Brain Abnormalities | - | Anterior intrasellar arachnoid cyst. | - | - | - | - | - |
| Hypotonia | + | + | - | - | + | - | - |
| Other | Severe motor impairment. | Ligamentous laxity. | - | Non-spastic muscle contractures. | Oppositional behaviors, hetero-aggressive attitudes with refusal of body contact. | Asperger, hearing loss, macrosomia. |
| Klein et al., 2005 [2] | Rauen et al., 2002 [7] | Schluth et al., 2008 [8] | McKenna et al., 2019 [4] | Niclass et al., 2020_1 [5] | Niclass et al., 2020_2 [5] | Oliveira et al., 2015 [6] | Matsumoto et al., 2014 [3] | Niclass et al., 2020_3 [5] | Cano et al., 2016 [1] | Akilapa et al., 2015 [9] | Lenk et al., 2018 [10] | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Growth retardation | + | + | + | |||||||||
| DD/ID | + | + | + | + | + | + | + | + | + | + | + | |
| Prominent forehead | + | + | + | + | + | + | + | + | ||||
| Hypertelorism | + | + | + | + | ||||||||
| Low set ears | + | + | + | + | + | + | + | + | ||||
| Short nose | + | + | + | + | + | + | + | |||||
| Other dysmorphisms | Anteverted nostrils, long philtrum, thin lips, microgna-thia | Anteverted nares | Long high palpebral fissures, hypoplastic nostrils | Flat face, hypoplastic nostrils, dysplastic left ear | Long philtrum, high arched palate | Upslanted palpebral fissures, anteverted nares, wide philtrum, thin upper lip, prominent chin | Microphthalmia, microdontia | Wide nasal base, low hanging columella, thin upper lip, wide mouth, down-slanting palpebral fissures | ||||
| 2–3 toe syndactyly + single palmar crease | + | + | + | + | + | + | ||||||
| Cardiac anomalies | + | + | + | + | ||||||||
| Ectodermal abnormalities | + | + | + | + | + | + | ||||||
| Ocular abnormalities (strabismus; hyperopia) | + | + | + | + | + | |||||||
| Genitourinary anomalies | + | + | + | + | ||||||||
| Brain abnormalities | + | + | + | + | + | + | ||||||
| Hypotonia | + | + | + | |||||||||
| Other Features | Hearing loss | Ataxia, dysarthria | ASD | Myopia, microcornea | Tall stature |
| Patients | 261,816 | 1582 | 1875 | 331,592 | 291,358 | 306,170 | 323,949 | 273,686 |
|---|---|---|---|---|---|---|---|---|
| Growth retardation | + | |||||||
| DD/ID | + | + | + | + | + | + | ||
| Prominent forehead | + | + | + | |||||
| Hypertelorism | ||||||||
| Low set ears | + | + | + | |||||
| Short nose | ||||||||
| Other dysmorphisms | Upslanted palpebral fissure, wide mouth | Macrocephaly, brachycephaly | Long philtrum, relative macrocephaly, short neck, upslanted palpebral fissures | Highly arched eyebrow, upslanted palpebral fissures | ||||
| 2–3 toe syndactyly + single palmar crease | + | |||||||
| Cardiac anomalies | + | |||||||
| Ectodermal abnormalities | + | + | ||||||
| Ocular abnormalities (strabismus; hyperopia) | + | |||||||
| Genitourinary anomalies | ||||||||
| Brain abnormalities | ||||||||
| Hypotonia | + | |||||||
| Other features | Joint laxity | Aggressive behavior, Peters anomaly | Corneal dystrophy, hand polydactyly, hypospadias |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Recalcati, M.P.; Catusi, I.; Garzo, M.; Redaelli, S.; Massimello, M.; Maitz, S.B.; Gentile, M.; Ponzi, E.; Orsini, P.; Zilio, A.; et al. 12q21 Interstitial Deletions: Seven New Syndromic Cases Detected by Array-CGH and Review of the Literature. Genes 2022, 13, 780. https://doi.org/10.3390/genes13050780
Recalcati MP, Catusi I, Garzo M, Redaelli S, Massimello M, Maitz SB, Gentile M, Ponzi E, Orsini P, Zilio A, et al. 12q21 Interstitial Deletions: Seven New Syndromic Cases Detected by Array-CGH and Review of the Literature. Genes. 2022; 13(5):780. https://doi.org/10.3390/genes13050780
Chicago/Turabian StyleRecalcati, Maria Paola, Ilaria Catusi, Maria Garzo, Serena Redaelli, Marta Massimello, Silvia Beatrice Maitz, Mattia Gentile, Emanuela Ponzi, Paola Orsini, Anna Zilio, and et al. 2022. "12q21 Interstitial Deletions: Seven New Syndromic Cases Detected by Array-CGH and Review of the Literature" Genes 13, no. 5: 780. https://doi.org/10.3390/genes13050780
APA StyleRecalcati, M. P., Catusi, I., Garzo, M., Redaelli, S., Massimello, M., Maitz, S. B., Gentile, M., Ponzi, E., Orsini, P., Zilio, A., Montaldi, A., Calò, A., Capra, A. P., Briuglia, S., La Rosa, M. A., Grillo, L., Romano, C., Bianca, S., Malacarne, M., ... Larizza, L. (2022). 12q21 Interstitial Deletions: Seven New Syndromic Cases Detected by Array-CGH and Review of the Literature. Genes, 13(5), 780. https://doi.org/10.3390/genes13050780