
Metagenomics Reveals the Diversity and Taxonomy of Carbohydrate-Active Enzymes and Antibiotic Resistance Genes in Suancai Bacterial Communities



Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Sequencing
2.2. Metagenome Assembly and Taxonomic Assignment
2.3. Functional Annotation
2.4. ARGs Identification
2.5. Statistical Analysis
3. Results and Discussion
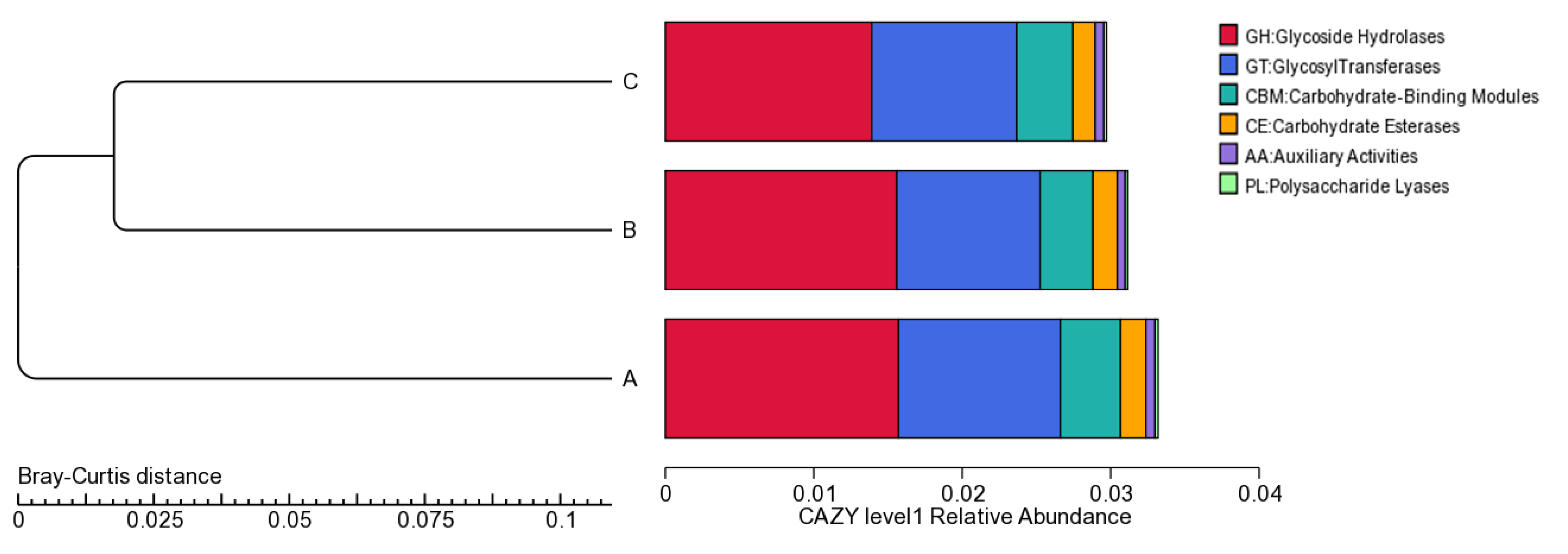
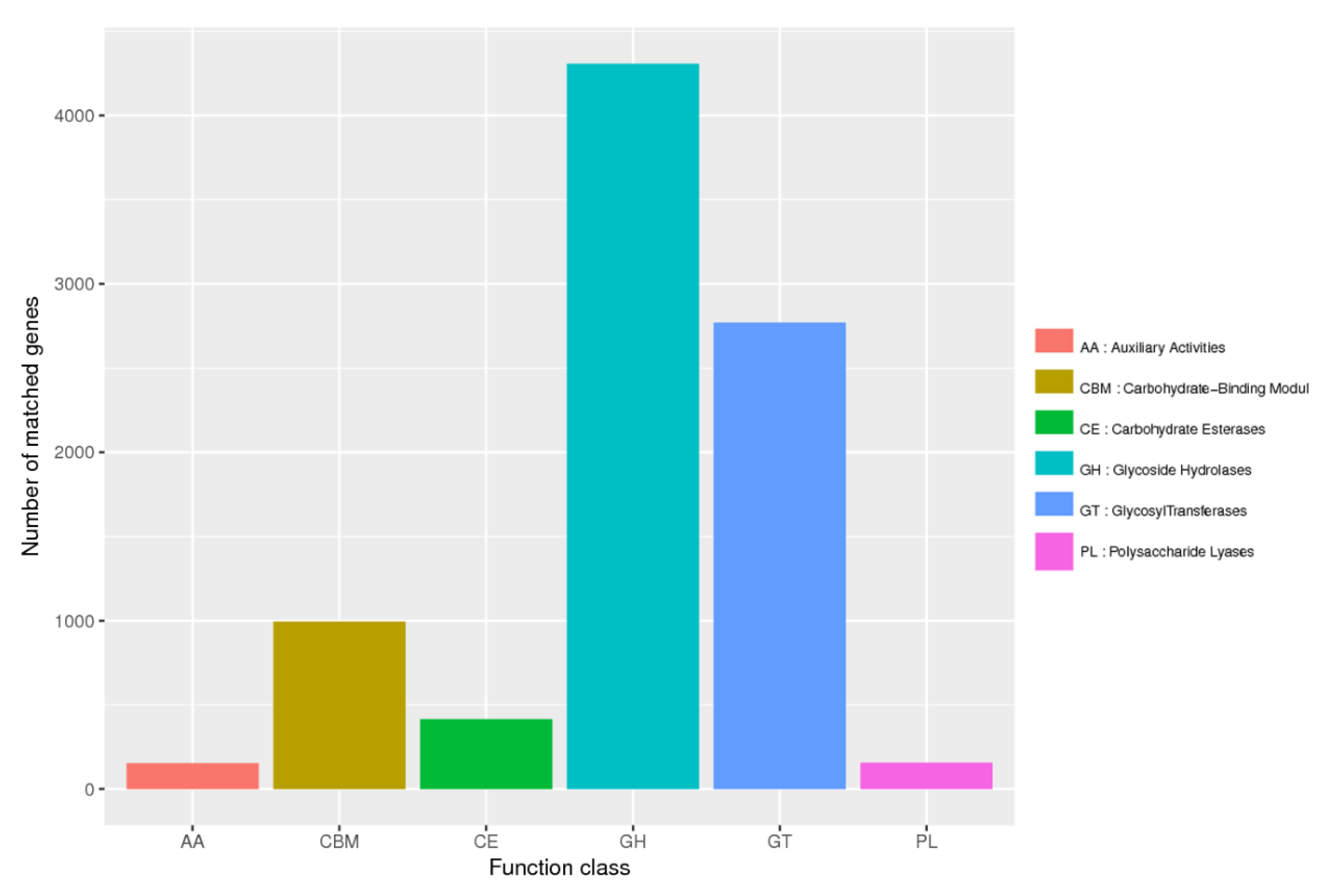
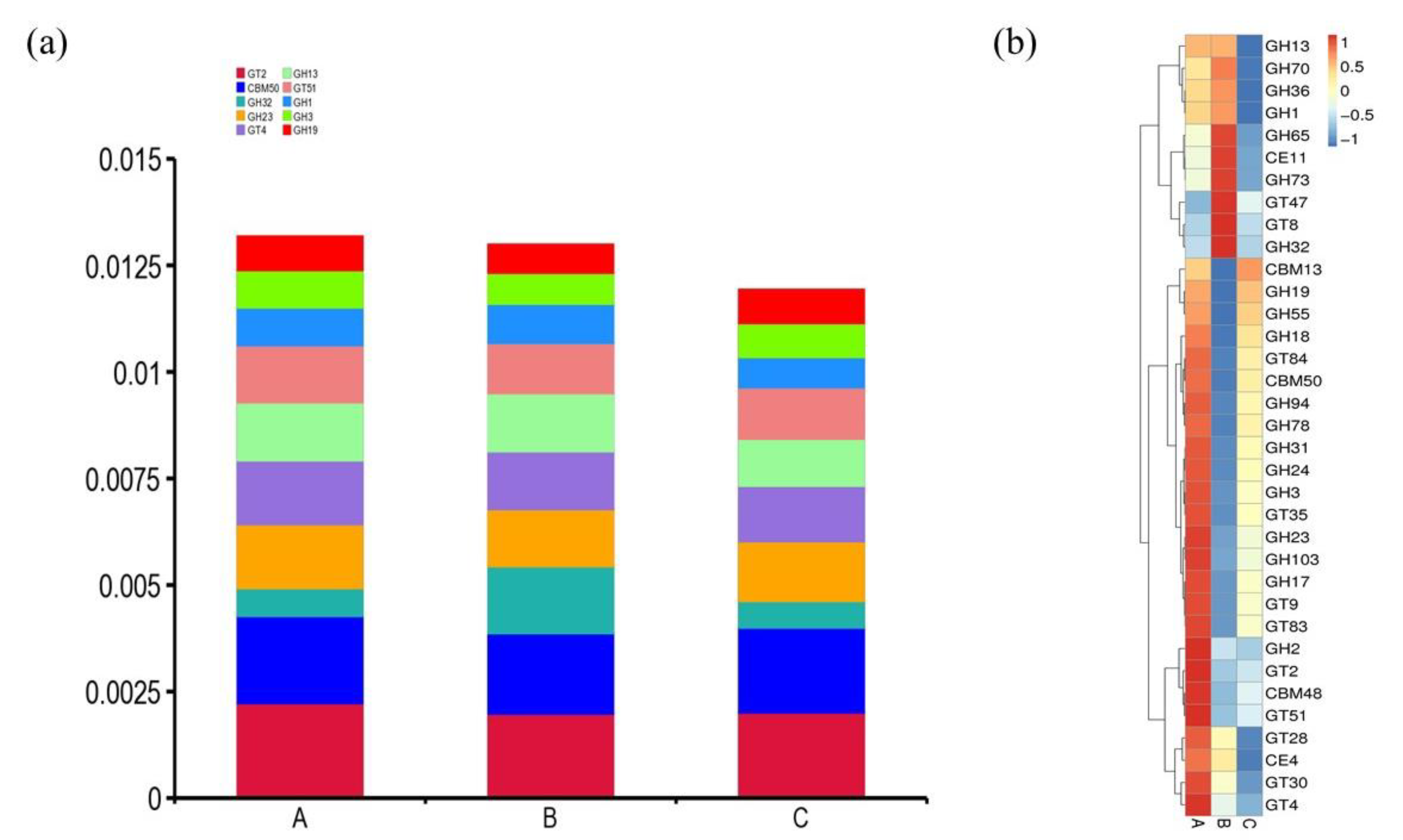
3.1. Metagenomic Assembly Revealed CAZymes
3.2. Phylogenetic Distribution of CAZymes
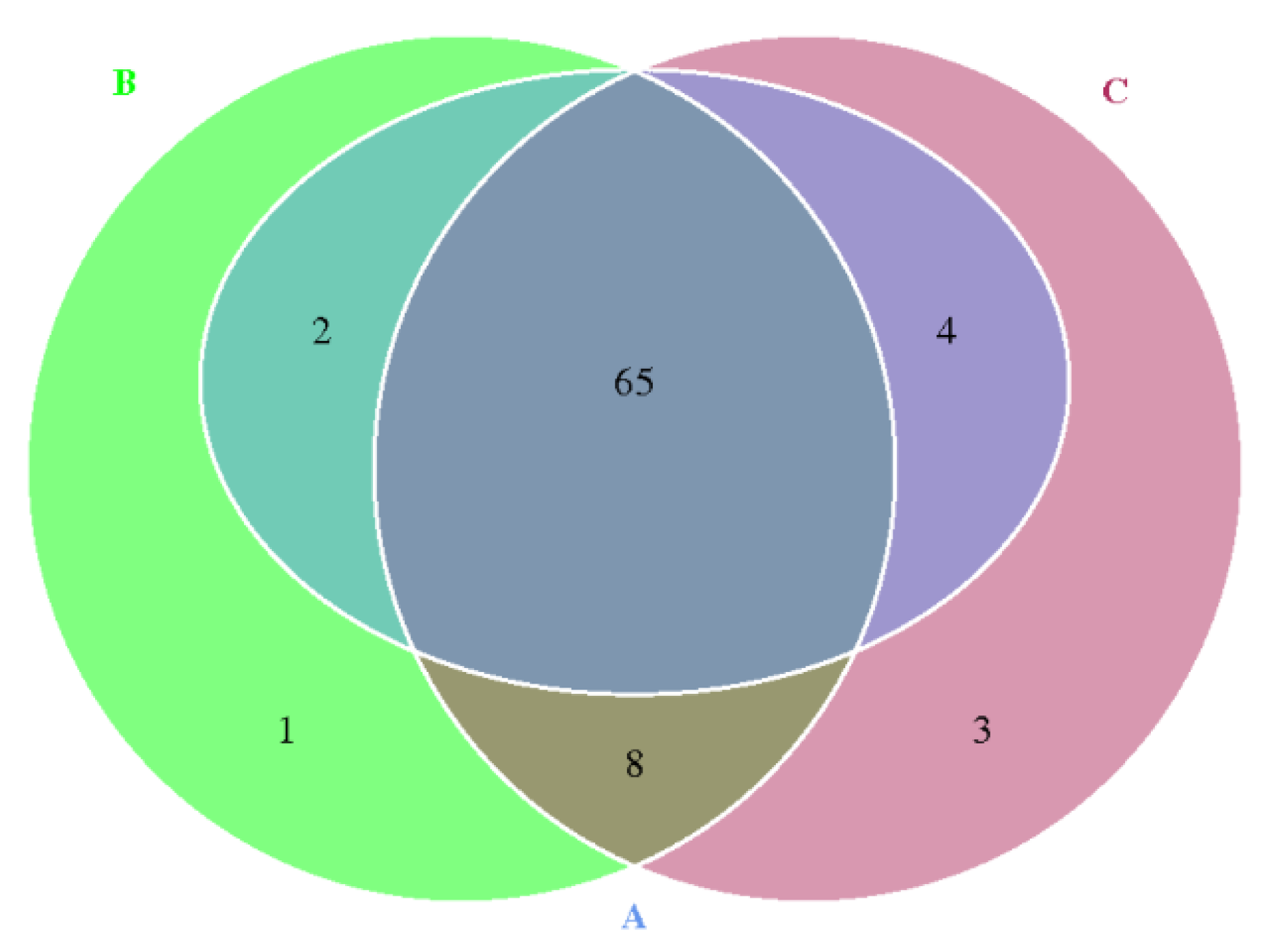
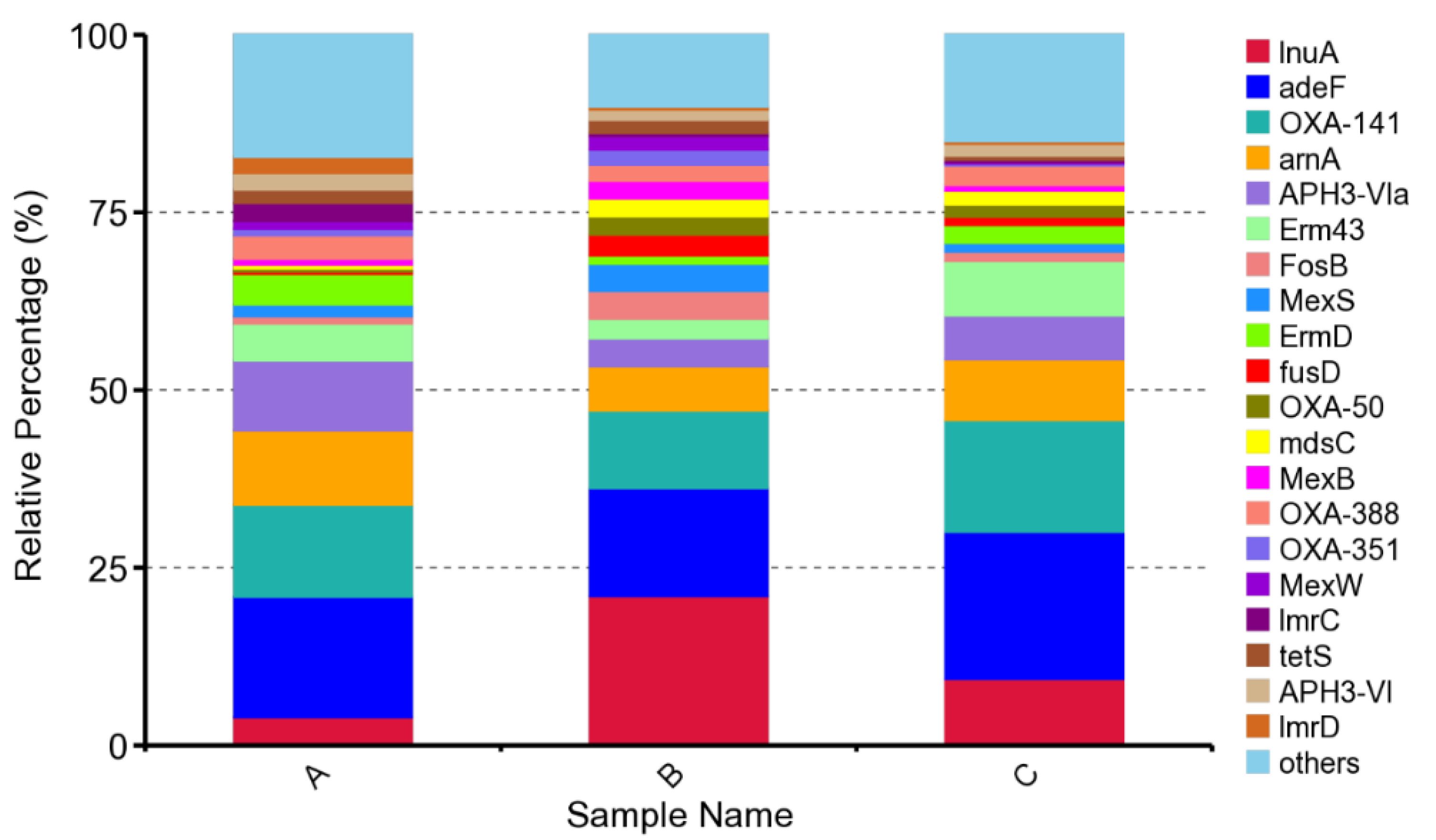
3.3. Occurrence and Characteristics of ARGs during Suancai Fermentation
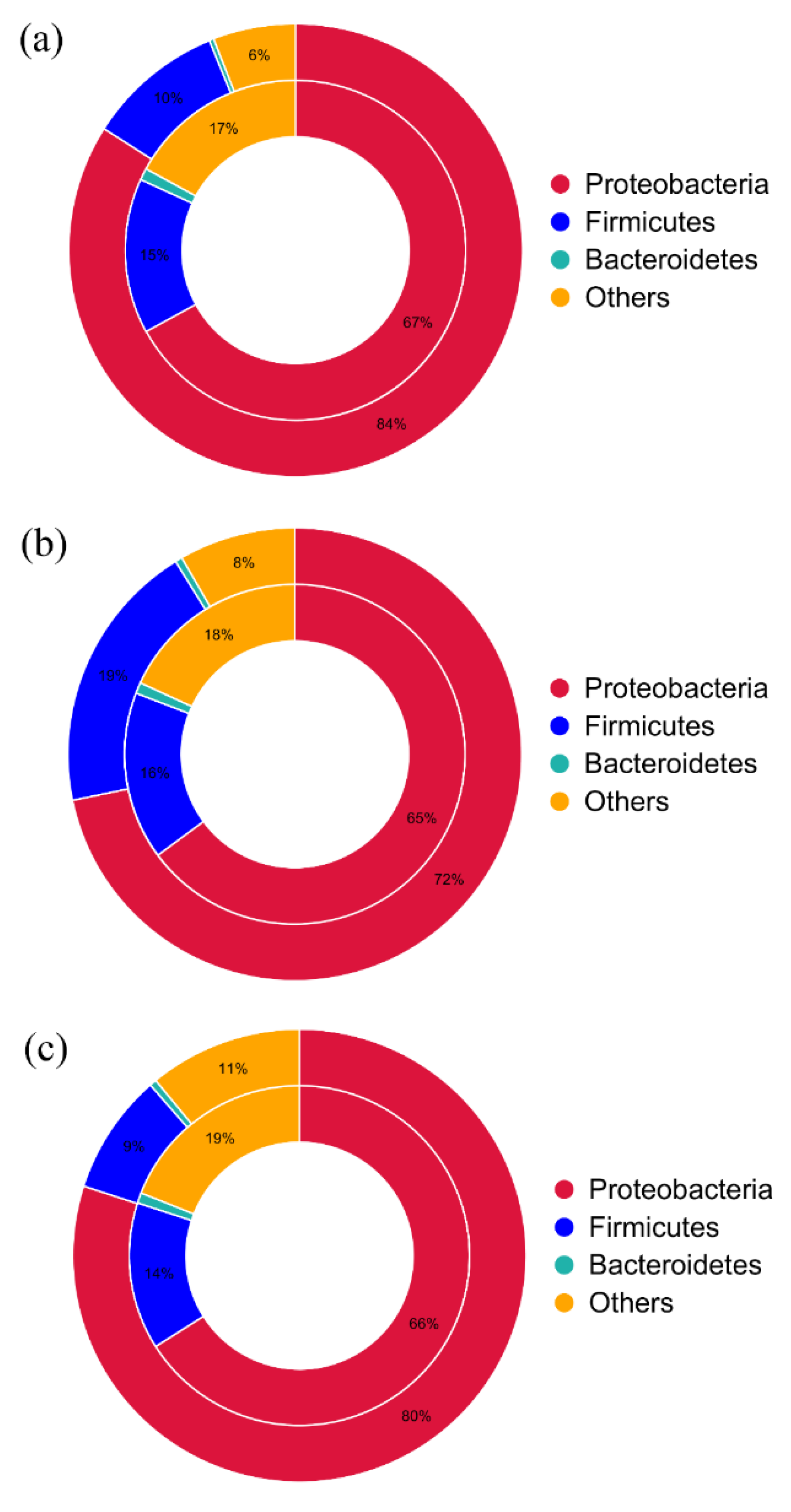
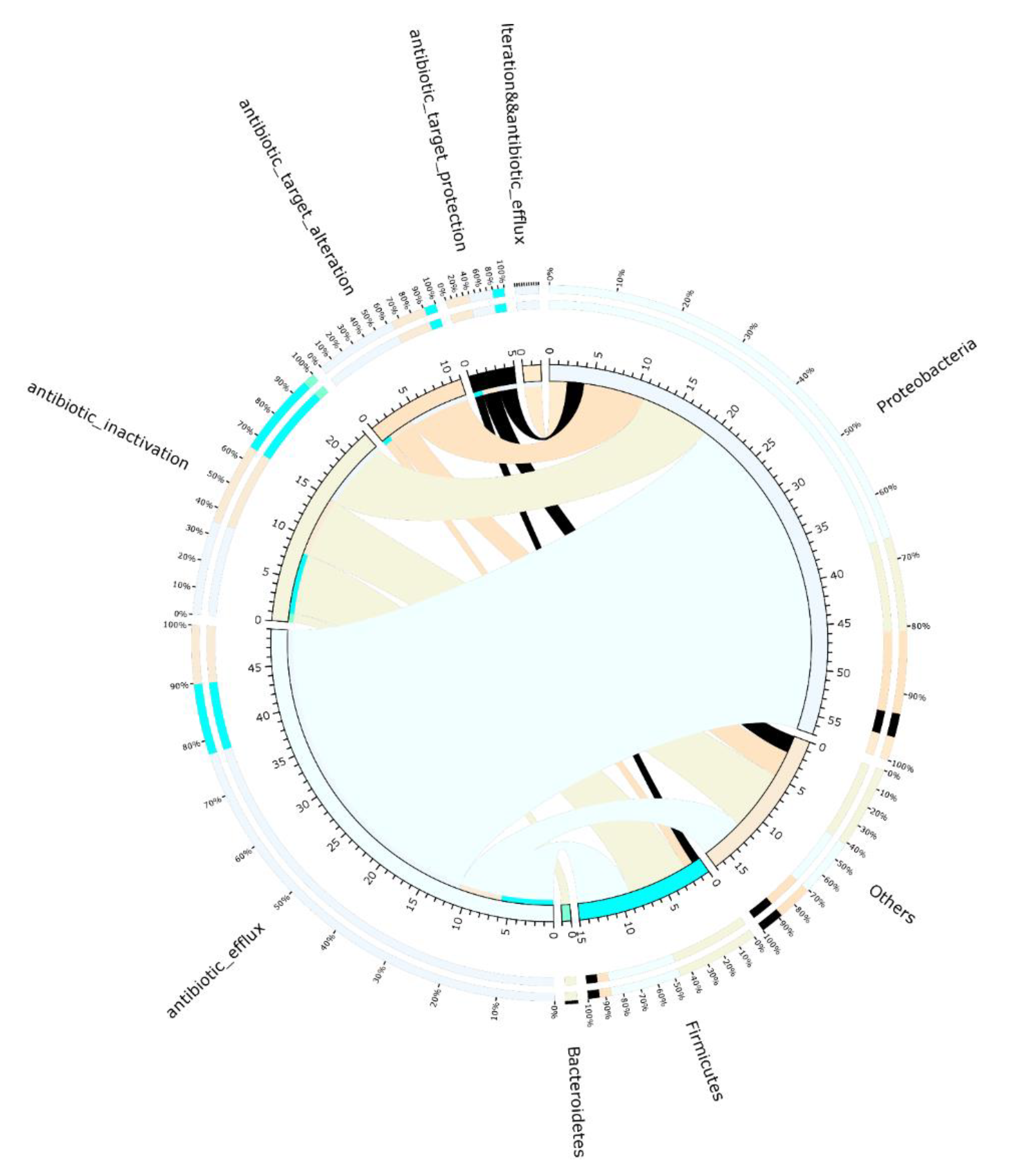
3.4. Correlation of ARGs and Their Potential Hosts
3.5. Resistance Mechanisms
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Selhub, E.M.; Logan, A.C.; Bested, A.C. Fermented foods, microbiota, and mental health: Ancient practice meets nutritional psychiatry. J. Physiol. Anthropol. 2014, 33, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rollan, G.C.; Gerez, C.L.; LeBlanc, J.G. Lactic Fermentation as a Strategy to Improve the Nutritional and Functional Values of Pseudocereals. Front. Nutr. 2019, 6, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, S.; Cachaldora, A.; Gomez, M.; Franco, I.; Carballo, J. Monitoring the bacterial population dynamics during the ripening of Galician chorizo, a traditional dry fermented Spanish sausage. Food Microbiol. 2013, 33, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Comunian, R.; Daga, E.; Dupre, I.; Paba, A.; Devirgiliis, C.; Piccioni, V.; Perozzi, G.; Zonenschain, D.; Rebecchi, A.; Morelli, L.; et al. Susceptibility to tetracycline and erythromycin of Lactobacillus paracasei strains isolated from traditional Italian fermented foods. Int. J. Food Microbiol. 2010, 138, 151–156. [Google Scholar] [CrossRef]
- Niccum, B.A.; Kastman, E.K.; Kfoury, N.; Robbat, A., Jr.; Wolfe, B.E. Strain-Level Diversity Impacts Cheese Rind Microbiome Assembly and Function. Msystems 2020, 5, e00149-20. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.M.; Macori, G.; Kilcawley, K.N.; Cotter, P.D. Meta-analysis of cheese microbiomes highlights contributions to multiple aspects of quality. Nat. Food 2020, 1, 500–510. [Google Scholar] [CrossRef]
- Lopez-Lopez, O.; Cerdan, M.E.; Gonzalez-Siso, M.I. Hot spring metagenomics. Life 2013, 3, 308–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amoutzias, G.D.; Chaliotis, A.; Mossialos, D. Discovery Strategies of Bioactive Compounds Synthesized by Nonribosomal Peptide Synthetases and Type-I Polyketide Synthases Derived from Marine Microbiomes. Mar. Drugs 2016, 14, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Tay, M.Y.F.; Aung, K.T.; Seow, K.L.G.; Ng, L.C.; Purbojati, R.W.; Drautz-Moses, D.I.; Schuster, S.C.; Schlundt, J. Phenotypic and genotypic characterization of antimicrobial resistant Escherichia coli isolated from ready-to-eat food in Singapore using disk diffusion, broth microdilution and whole genome sequencing methods. Food Control 2019, 99, 89–97. [Google Scholar] [CrossRef]
- Li, B.; Yang, Y.; Ma, L.; Ju, F.; Guo, F.; Tiedje, J.M.; Zhang, T. Metagenomic and network analysis reveal wide distribution and co-occurrence of environmental antibiotic resistance genes. Isme J. 2015, 9, 2490–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanderson, H.; Fricker, C.; Brown, R.S.; Majury, A.; Liss, S.N. Antibiotic resistance genes as an emerging environmental contaminant. Environ. Rev. 2016, 24, 205–218. [Google Scholar] [CrossRef]
- Li, L.-G.; Xia, Y.; Zhang, T. Co-occurrence of antibiotic and metal resistance genes revealed in complete genome collection. Isme J. 2017, 11, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Kirbis, A.; Krizman, M. Spread of Antibiotic Resistant Bacteria from Food of Animal Origin to Humans and vice Versa. In Proceedings of the 58th International Meat Industry Conference (MeatCon), Belgrade, Serbia, 4–7 October 2015; pp. 148–151. [Google Scholar]
- Li, Y.; Cao, W.; Liang, S.; Yamasaki, S.; Chen, X.; Shi, L.; Ye, L. Metagenomic characterization of bacterial community and antibiotic resistance genes in representative ready-to-eat food in southern China. Sci. Rep. 2020, 10, 15175. [Google Scholar] [CrossRef] [PubMed]
- Founou, L.L.; Founou, R.C.; Essack, S.Y. Antibiotic Resistance in the Food Chain: A Developing Country-Perspective. Front. Microbiol. 2016, 7, 1881. [Google Scholar] [CrossRef] [PubMed]
- Rolain, J.-M. Food and human gut as reservoirs of transferable antibiotic resistance encoding genes. Front. Microbiol. 2013, 4, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, G.; Hu, M.; Li, X.; Pan, Z.; Li, M.; Li, L.; Zheng, Z.; Yang, M. Metagenomics reveals the diversity and taxonomy of antibiotic resistance genes in sufu bacterial communities. Food Control 2021, 121, 107641. [Google Scholar] [CrossRef]
- Abriouel, H.; Knapp, C.; Gálvez, A.; Benomar, N. Antibiotic resistance profile of microbes from traditional fermented foods. In Fermented Foods in Health and disease Prevention; Elsevier: Amsterdam, The Netherlands, 2017; pp. 675–704. [Google Scholar]
- Martínez, J.; Coque, T.M.; Baquero, F. What is a resistance gene? Ranking risk in resistomes. Nat. Rev. Microbiol. 2015, 13, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Capita, R.; Alonso-Calleja, C. Antibiotic-Resistant Bacteria: A Challenge for the Food Industry. Crit. Rev. Food Sci. Nutr. 2013, 53, 11–48. [Google Scholar] [CrossRef]
- Aarts, H.; Margolles, A. Antibiotic resistance genes in food and gut (non-pathogenic) bacteria. Bad genes in good bugs. Front. Microbiol. 2015, 5, 754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Hoek, A.H.A.M.; Mevius, D.; Guerra, B.; Mullany, P.; Roberts, A.P.; Aarts, H.J.M. Acquired antibiotic resistance genes: An overview. Front. Microbiol. 2011, 2, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.H.; Manuzon, M.; Lehman, M.; Wan, K.; Luo, H.L.; Wittum, T.E.; Yousef, A.; Bakaletz, L.O. Food commensal microbes as a potentially important avenue in transmitting antibiotic resistance genes. FEMS Microbiol. Lett. 2006, 254, 226–231. [Google Scholar] [CrossRef]
- Doster, E.; Thomas, K.M.; Weinroth, M.D.; Parker, J.K.; Crone, K.K.; Arthur, T.M.; Schmidt, J.W.; Wheeler, T.L.; Belk, K.E.; Morley, P.S. Metagenomic Characterization of the Microbiome and Resistome of Retail Ground Beef Products. Front. Microbiol. 2020, 11, 541972. [Google Scholar] [CrossRef]
- Zhao, S.; Blickenstaff, K.; Bodeis-Jones, S.; Gaines, S.A.; Tong, E.; McDermott, P.F. Comparison of the Prevalences and Antimicrobial Resistances of Escherichia coli Isolates from Different Retail Meats in the United States, 2002 to 2008. Appl. Environ. Microbiol. 2012, 78, 1701–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, L.; Sun, Y.; Shi, L.; Yan, H. Characterization of antimicrobial resistance genes and class 1 integrase gene in raw meat and aquatic product, fresh vegetable and fruit, and swine manure in southern China. Food Control 2019, 104, 240–246. [Google Scholar] [CrossRef]
- Cao, Y.; Fanning, S.; Proos, S.; Jordan, K.; Srikumar, S. A Review on the Applications of Next Generation Sequencing Technologies as Applied to Food-Related Microbiome Studies. Front. Microbiol. 2017, 8, 1829. [Google Scholar] [CrossRef] [PubMed]
- Kanger, K.; Guilford, N.G.H.; Lee, H.; Nesbo, C.L.; Truu, J.; Edwards, E.A. Antibiotic resistome and microbial community structure during anaerobic co-digestion of food waste, paper and cardboard. Fems Microbiol. Ecol. 2020, 96, fiaa006. [Google Scholar] [CrossRef]
- Sommer, M.O.A.; Dantas, G.; Church, G.M. Functional Characterization of the Antibiotic Resistance Reservoir in the Human Microflora. Science 2009, 325, 1128–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengtsson-Palme, J.; Kristiansson, E.; Larsson, D.G.J. Environmental factors influencing the development and spread of antibiotic resistance. Fems Microbiol. Rev. 2018, 42, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Bibi, F.; Hashem, A.M.; Azhar, E.I. Comparative metagenomics and characterization of antimicrobial resistance genes in pasteurized and homemade fermented Arabian laban. Food Res. Int. 2020, 137, 109639. [Google Scholar] [CrossRef]
- Segata, N.; Waldron, L.; Ballarini, A.; Narasimhan, V.; Jousson, O.; Huttenhower, C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 2012, 9, 811–814. [Google Scholar] [CrossRef]
- Song, Q.; Zhao, F.; Wang, B.; Han, Y.; Zhou, Z. Metagenomic insights into Chinese northeast suancai: Predominance and diversity of genes associated with nitrogen metabolism in traditional household suancai fermentation. Food Res. Int. 2021, 139, 109924. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler (vol 1, 18, 2012). Gigascience 2015, 4, 2047-217X. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villar, E.; Farrant, G.K.; Follows, M.; Garczarek, L.; Speich, S.; Audic, S.; Bittner, L.; Blanke, B.; Brum, J.R.; Brunet, C.; et al. Environmental characteristics of Agulhas rings affect interocean plankton transport. Science 2015, 348, 1261447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotillard, A.; Kennedy, S.P.; Kong, L.C.; Prifti, E.; Pons, N.; Le Chatelier, E.; Almeida, M.; Quinquis, B.; Levenez, F.; Galleron, N.; et al. Dietary intervention impact on gut microbial gene richness. Nature 2013, 500, 585–588. [Google Scholar] [CrossRef]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef] [Green Version]
- Powell, S.; Forslund, K.; Szklarczyk, D.; Trachana, K.; Roth, A.; Huerta-Cepas, J.; Gabaldon, T.; Rattei, T.; Creevey, C.; Kuhn, M.; et al. eggNOG v4.0: Nested orthology inference across 3686 organisms. Nucleic Acids Res. 2014, 42, D231–D239. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z.; et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun. 2015, 6, 6528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef] [PubMed]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The Comprehensive Antibiotic Resistance Database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, J.R.; Curtis, J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 326–349. [Google Scholar] [CrossRef]
- Schmid, J.; Heider, D.; Wendel, N.J.; Sperl, N.; Sieber, V. Bacterial Glycosyltransferases: Challenges and Opportunities of a Highly Diverse Enzyme Class Toward Tailoring Natural Products. Front. Microbiol. 2016, 7, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svartstrom, O.; Alneberg, J.; Terrapon, N.; Lombard, V.; de Bruijn, I.; Malmsten, J.; Dalin, A.-M.; El Muller, E.; Shah, P.; Wilmes, P.; et al. Ninety-nine de novo assembled genomes from the moose (Alces alces) rumen microbiome provide new insights into microbial plant biomass degradation. Isme J. 2017, 11, 2538–2551. [Google Scholar] [CrossRef] [PubMed]
- Viridiana, C.R.; Lidia, D.A.; Audry, P.L.; Humberto, H.S. Lactic Acid Bacteria Isolated from Vegetable Fermentations: Probiotic Characteristics; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Pan, L.; Han, Y.; Zhou, Z. In vitro prebiotic activities of exopolysaccharide from Leuconostoc pseudomesenteroides XG5 and its effect on the gut microbiota of mice. J. Funct. Foods 2020, 67, 103853. [Google Scholar] [CrossRef]
- Leech, J.; Cabrera-Rubio, R.; Walsh, A.M.; Macori, G.; Walsh, C.J.; Barton, W.; Finnegan, L.; Crispie, F.; O’Sullivan, O.; Claesson, M.J.; et al. Fermented-Food Metagenomics Reveals Substrate-Associated Differences in Taxonomy and Health-Associated and Antibiotic Resistance Determinants. Msystems 2020, 5, e00522-20. [Google Scholar] [CrossRef] [PubMed]
- Hachler, H.; Santanam, P.; Kayser, F.H. Sequence and characterization of a novel chromosomal aminoglycoside phosphotransferase gene, aph (3’)-IIb, in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1996, 40, 1254–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, L.; Jin, S.G. aph(3 ‘)-IIb, a gene encoding an aminoglycoside-modifying enzyme, is under the positive control of surrogate regulator HpaA. Antimicrob. Agents Chemother. 2003, 47, 3867–3876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Mima, T.; Komori, Y.; Morita, Y.; Kuroda, T.; Mizushima, T.; Tsuchiya, T. A new member of the tripartite multidrug efflux pumps, MexVW-OprM, in Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2003, 52, 572–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfaresi, M. Whole Genome Sequencing of Klebsiella pneumoniae Strain Unravels a New Model for the Development of Extensive Drug Resistance in Enterobacteriaceae. Open Microbiol. J. 2018, 12, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Toth, A.G.; Csabai, I.; Maroti, G.; Jerzsele, A.; Dubecz, A.; Patai, A.V.; Judge, M.F.; Nagy, S.A.; Makrai, L.; Banyai, K.; et al. A glimpse of antimicrobial resistance gene diversity in kefir and yoghurt. Sci. Rep. 2020, 10, 22458. [Google Scholar] [CrossRef] [PubMed]
- Blokesch, M. Natural competence for transformation. Curr. Biol. 2016, 26, R1126–R1130. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | ARG Type | Resistance Mechanism | Abundance in Sample A | Abundance in Sample B | Abundance in Sample C |
|---|---|---|---|---|---|
| lnuA | lincosamide | antibiotic inactivation× | 1.53 × 10−5 | 0.000109 | 3.41 × 10−5 |
| adeF | Multidrug | antibiotic efflux | 6.55 × 10−5 | 7.86 × 10−5 | 7.56 × 10−5 |
| OXA−141 | Multidrug | antibiotic inactivation | 5.00 × 10−5 | 5.67 × 10−5 | 5.74 × 10−5 |
| arnA | peptide | antibiotic target alteration | 4.03 × 10−5 | 3.20 × 10−5 | 3.12 × 10−5 |
| APH3-VIa | aminoglycoside | antibiotic inactivation | 3.80 × 10−5 | 2.05 × 10−5 | 2.26 × 10−5 |
| Erm43 | Multidrug | antibiotic target alteration | 2.01 × 10−5 | 1.42 × 10−5 | 2.80 × 10−5 |
| FosB | fosfomycin | antibiotic inactivation | 3.83 × 10−6 | 2.02 × 10−5 | 4.69 × 10−6 |
| MexS | Multidrug | antibiotic efflux | 6.48 × 10−6 | 2.00 × 10−5 | 4.52 × 10−6 |
| ErmD | Multidrug | antibiotic target alteration | 1.66 × 10−5 | 6.00 × 10−6 | 9.15 × 10−6 |
| fusD | fusidic acid | antibiotic inactivation | 1.39 × 10−6 | 1.51 × 10−5 | 4.27 × 10−6 |
| OXA-50 | Multidrug | antibiotic inactivation | 1.50 × 10−6 | 1.33 × 10−5 | 6.17 × 10−6 |
| mdsC | Multidrug | antibiotic efflux | 2.09 × 10−6 | 1.30 × 10−5 | 7.23 × 10−6 |
| MexB | Multidrug | antibiotic efflux | 3.21 × 10−6 | 1.29 × 10−5 | 2.87 × 10−6 |
| OXA-388 | Multidrug | antibiotic inactivation | 1.28 × 10−5 | 1.16 × 10−5 | 1.00 × 10−5 |
| OXA-351 | Multidrug | antibiotic inactivation | 3.55 × 10−6 | 1.11 × 10−5 | 9.50 × 10−7 |
| MexW | Multidrug | resistance-nodulation-cell division (RND) antibiotic efflux pump | 4.24 × 10−6 | 9.82 × 10−6 | 6.88 × 10−07 |
| lmrC | lincosamide | antibiotic efflux | 9.78 × 10−6 | 2.23 × 10−6 | 1.39 × 10−6 |
| tetS | tetracycline | antibiotic target protection | 7.24 × 10−6 | 9.75 × 10−6 | 1.94 × 10−6 |
| APH3-VI | aminoglycoside | antibiotic inactivation | 8.98 × 10−6 | 7.35 × 10−6 | 6.19 × 10−6 |
| lmrD | lincosamide | antibiotic efflux | 8.87 × 10−6 | 1.96 × 10−6 | 1.27 × 10−6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Q.; Wang, B.; Han, Y.; Zhou, Z. Metagenomics Reveals the Diversity and Taxonomy of Carbohydrate-Active Enzymes and Antibiotic Resistance Genes in Suancai Bacterial Communities. Genes 2022, 13, 773. https://doi.org/10.3390/genes13050773
Song Q, Wang B, Han Y, Zhou Z. Metagenomics Reveals the Diversity and Taxonomy of Carbohydrate-Active Enzymes and Antibiotic Resistance Genes in Suancai Bacterial Communities. Genes. 2022; 13(5):773. https://doi.org/10.3390/genes13050773
Chicago/Turabian StyleSong, Qiaozhi, Binbin Wang, Ye Han, and Zhijiang Zhou. 2022. "Metagenomics Reveals the Diversity and Taxonomy of Carbohydrate-Active Enzymes and Antibiotic Resistance Genes in Suancai Bacterial Communities" Genes 13, no. 5: 773. https://doi.org/10.3390/genes13050773
APA StyleSong, Q., Wang, B., Han, Y., & Zhou, Z. (2022). Metagenomics Reveals the Diversity and Taxonomy of Carbohydrate-Active Enzymes and Antibiotic Resistance Genes in Suancai Bacterial Communities. Genes, 13(5), 773. https://doi.org/10.3390/genes13050773