
Reduction of BMPR2 mRNA Expression in Peripheral Blood of Pulmonary Arterial Hypertension Patients: A Marker for Disease Severity?

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
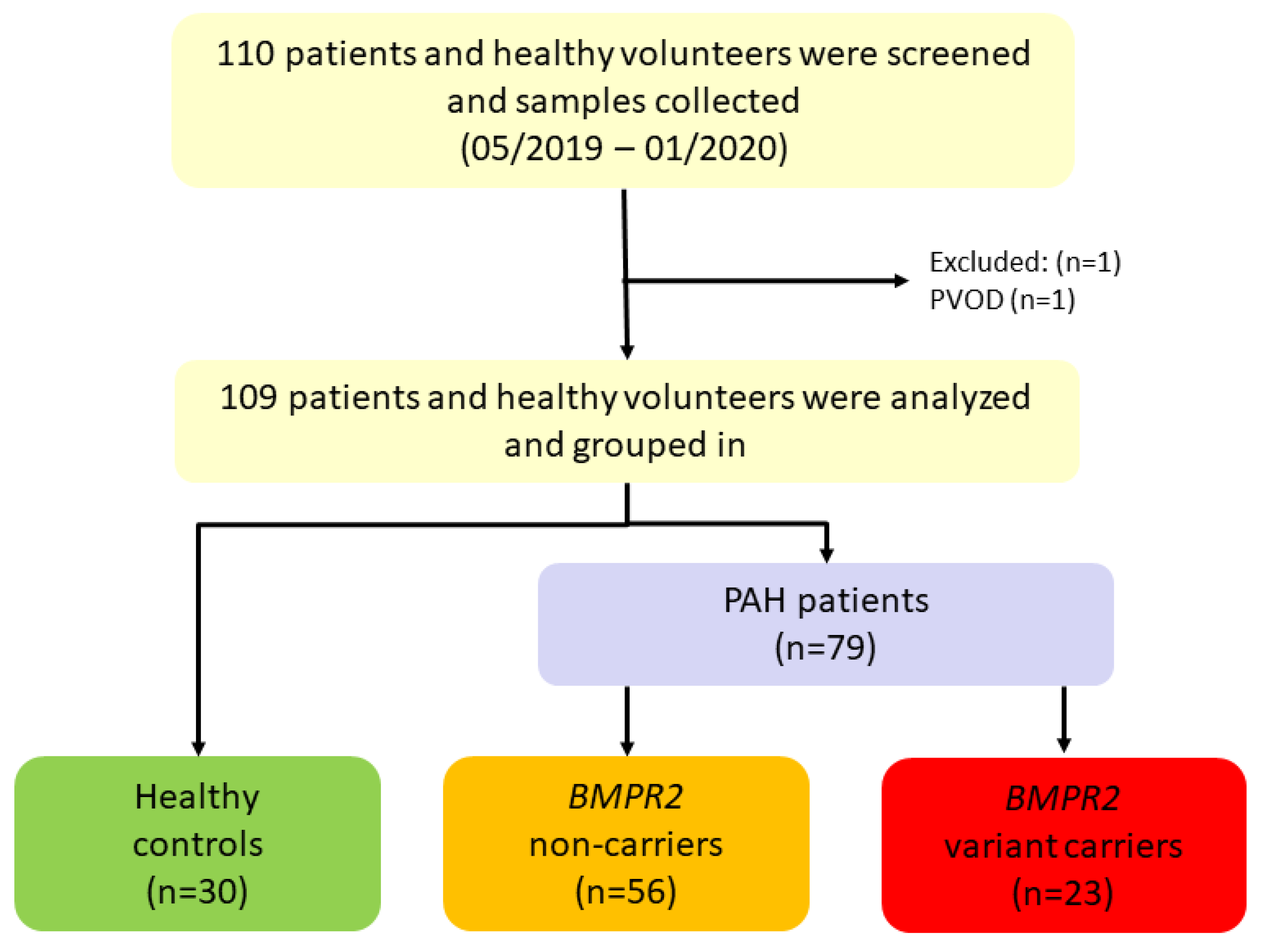
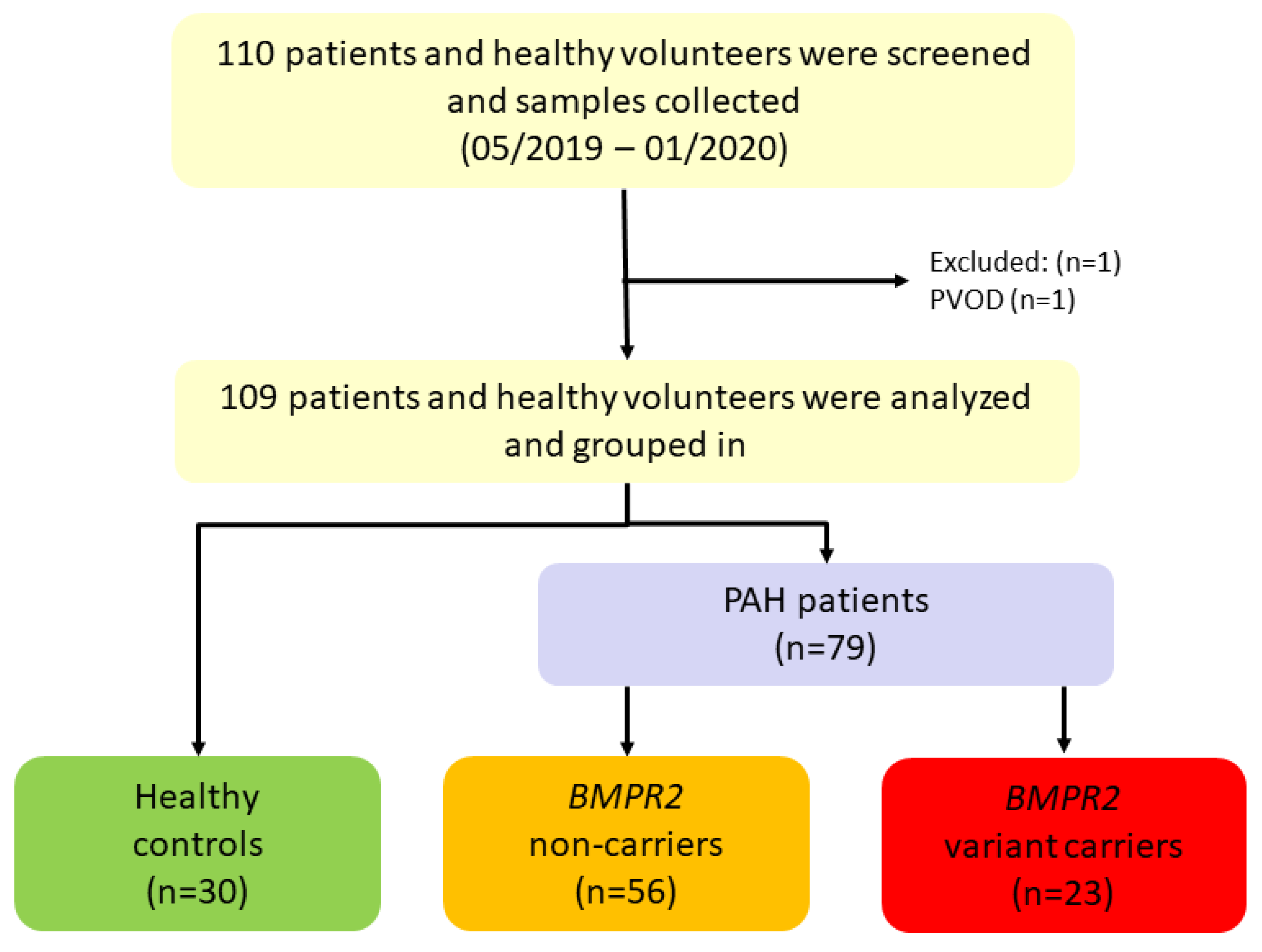
2.1. Study Design
2.2. Clinical Investigations
2.3. Genetic Investigations
2.4. Expression Level Analyses
2.5. Statistical Methods
3. Results
3.1. Baseline Characteristics
3.2. Clinical Differences between BMPR2 Variant Carriers and Non-Carriers
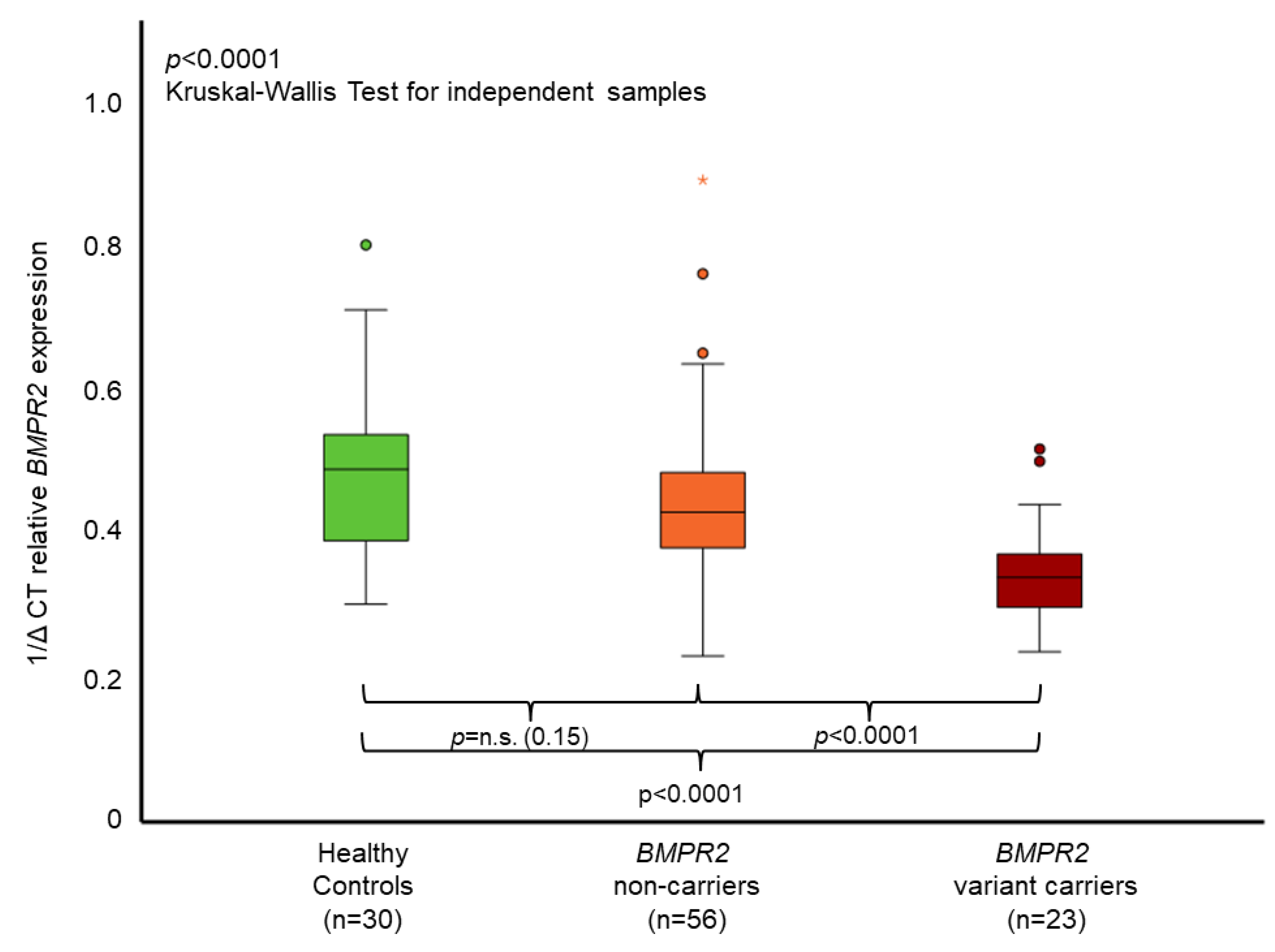
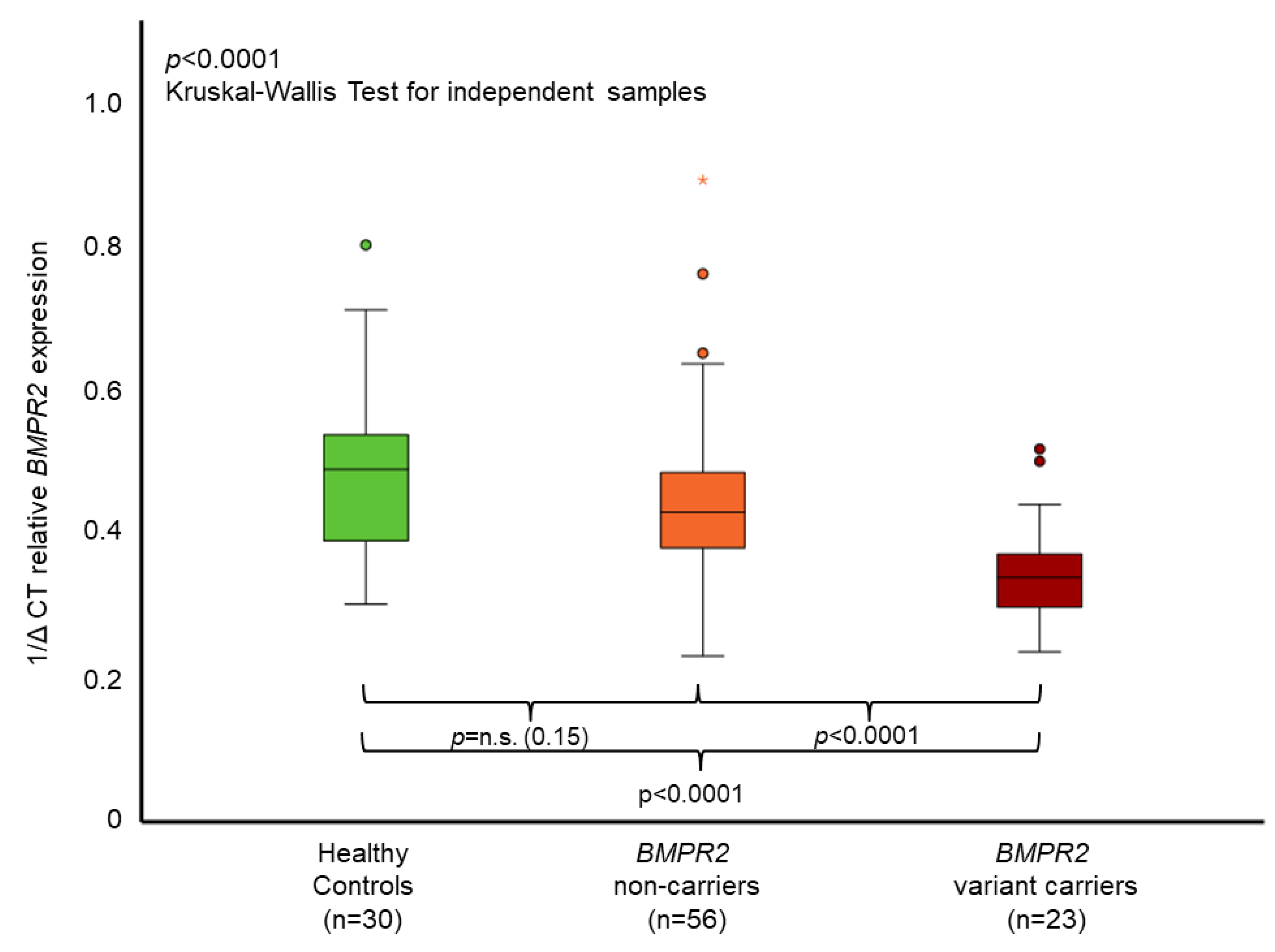
3.3. BMPR2 Characterization and Expression
3.4. Correlation of BMPR2 mRNA Expression with Further Laboratory and Clinical Characteristics
4. Discussion
4.1. Correlation of BMPR2 mRNA Expression with Further Laboratory and Clinical Characteristics
4.2. BMPR2 mRNA Expression and Correlation to Other Parameters
4.3. Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galiè, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [PubMed]
- Song, J.; Eichstaedt, C.A.; Rodríguez Viales, R.; Benjamin, N.; Harutyunova, S.; Fischer, C.; Grünig, E.; Hinderhofer, K. Identification of genetic defects in pulmonary arterial hypertension by a new gene panel diagnostic tool. Clin. Sci. 2016, 130, 2043–2052. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.D.; Eickelberg, O.; Elliott, C.G.; Geraci, M.W.; Hanaoka, M.; Loyd, J.E.; Newman, J.H.; Phillips, J.A., 3rd; Soubrier, F.; Trembath, R.C.; et al. Genetics and genomics of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2009, 54, S32–S42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfarr, N.; Szamalek-Hoegel, J.; Fischer, C.; Hinderhofer, K.; Nagel, C.; Ehlken, N.; Tiede, H.; Olschewski, H.; Reichenberger, F.; Ghofrani, A.H.; et al. Hemodynamic and clinical onset in patients with hereditary pulmonary arterial hypertension and BMPR2 mutations. Respir. Res. 2011, 12, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichstaedt, C.A.; Song, J.; Benjamin, N.; Harutyunova, S.; Fischer, C.; Grünig, E.; Hinderhofer, K. EIF2AK4 mutation as “second hit” in hereditary pulmonary arterial hypertension. Respir. Res. 2016, 17, 141. [Google Scholar] [PubMed] [Green Version]
- Atkinson, C.; Stewart, S.; Upton, P.D.; Machado, R.; Thomson, J.R.; Trembath, R.C.; Morrell, N.W. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 2002, 105, 1672–1678. [Google Scholar] [CrossRef] [Green Version]
- Jonigk, D.; Golpon, H.; Bockmeyer, C.L.; Maegel, L.; Hoeper, M.M.; Gottlieb, J.; Nickel, N.; Hussein, K.; Maus, U.; Lehmann, U.; et al. Plexiform lesions in pulmonary arterial hypertension composition, architecture, and microenvironment. Am. J. Pathol. 2011, 179, 167–179. [Google Scholar] [CrossRef]
- Evans, J.D.W.; Girerd, B.; Montani, D.; Wang, X.-J.; Galiè, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grünig, E.; Yan, Y.; et al. BMPR2 mutations and survival in pulmonary arterial hypertension: An individual participant data meta-analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Hinderhofer, K.; Fischer, C.; Pfarr, N.; Szamalek-Hoegel, J.; Lichtblau, M.; Nagel, C.; Egenlauf, B.; Ehlken, N.; Grünig, E. Identification of a new intronic BMPR2-mutation and early diagnosis of heritable pulmonary arterial hypertension in a large family with mean clinical follow-up of 12 years. PLoS ONE 2014, 9, e91374. [Google Scholar] [CrossRef] [PubMed]
- Montani, D.; Girerd, B.; Jaïs, X.; Laveneziana, P.; Lau, E.M.T.; Bouchachi, A.; Hascoët, S.; Günther, S.; Godinas, L.; Parent, F.; et al. Screening for pulmonary arterial hypertension in adults carrying a BMPR2 mutation. Eur. Respir. J. 2021, 58, 2004229. [Google Scholar] [CrossRef] [PubMed]
- Eichstaedt, C.A.; Verweyen, J.; Halank, M.; Benjamin, N.; Fischer, C.; Mayer, E.; Guth, S.; Wiedenroth, C.B.; Egenlauf, B.; Harutyunova, S.; et al. Myeloproliferative Diseases as Possible Risk Factor for Development of Chronic Thromboembolic Pulmonary Hypertension-A Genetic Study. Int. J. Mol. Sci. 2020, 21, 3339. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, M.A.; Granzow, M.; Warth, A.; Schnabel, P.A.; Thomas, M.; Herth, F.J.; Dienemann, H.; Muley, T.; Meister, M. Glycodelin: A New Biomarker with Immunomodulatory Functions in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2015, 21, 3529–3540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiekerkoetter, E.; Sung, Y.K.; Sudheendra, D.; Scott, V.; Del Rosario, P.; Bill, M.; Haddad, F.; Long-Boyle, J.; Hedlin, H.; Zamanian, R.T. Randomised placebo-controlled safety and tolerability trial of FK506 (tacrolimus) for pulmonary arterial hypertension. Eur. Respir. J. 2017, 50, 1602449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, M.; McLaughlin, V.; Gibbs, J.S.R.; Gomberg-Maitland, M.; Hoeper, M.M.; Preston, I.R.; Souza, R.; Waxman, A.; Escribano Subias, P.; Feldman, J.; et al. Sotatercept for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 384, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Eichstaedt, C.A.; Sassmannshausen, Z.; Shaukat, M.; Cao, D.; Xanthouli, P.; Gall, H.; Sommer, N.; Ghofrani, H.A.; Seyfarth, H.J.; Lerche, M.; et al. Gene panel diagnostics reveals new pathogenic variants in pulmonary arterial hypertension. Respir. Res. 2022, 23, 74. [Google Scholar] [CrossRef] [PubMed]
- Hemnes, A.R.; Humbert, M. Pathobiology of pulmonary arterial hypertension: Understanding the roads less travelled. Eur. Respir. Rev. 2017, 26, 170093. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| All Patients Mean ± SD or n and (%) n = 79 | n # | BMPR2 Non-Carriers Mean ± SD or n and (%) n = 56 | n # | BMPR2 Variant-Carriers Mean ± SD or n and (%) n = 23 | n # | p-Value ° | ||
|---|---|---|---|---|---|---|---|---|
| Characteristics | ||||||||
| Age, years | 51.4 ± 16.0 | 51.7 ± 16.6 | 50.9 ± 14.7 | 0.90 | ||||
| Height, cm | 168.5 ± 8.8 | 78 | 168.3 ± 9.1 | 55 | 169.0 ± 8.2 | 0.86 | ||
| Weight, kg | 74.0 ± 16.8 | 78 | 73.8 ± 18.0 | 55 | 74.5 ± 13.8 | 0.57 | ||
| Body mass index, kg/m2 | 26.0 ± 5.6 | 78 | 26.0 ± 6.0 | 55 | 26.0 ± 4.4 | 0.57 | ||
| Sex, female | 58 (73.4) | 41 (73.2) | 17 (73.9) | |||||
| Age at diagnosis, years | 42.0 ± 23.5 | 76 | 45.9 ± 17.8 | 31.0 ± 33.1 | 20 | 0.09 | ||
| Laboratory | ||||||||
| CRP, mg/L | 7.01 ± 9.08 | 76 | 6.61 ± 7.25 | 8.15 ± 13.11 | 20 | 0.43 | ||
| Hematocrit, L/L | 0.42 ± 0.04 | 0.42 ± 0.04 | 0.44 ± 0.04 | 0.11 | ||||
| Interleukin-6, pg/mL | 2.59 ± 3.00 | 71 | 2.76 ± 3.39 | 55 | 2.01 ± 0.05 | 16 | 0.07 | |
| Creatinine, mg/dL | 1.12 ± 2.50 | 1.23 ± 2.97 | 0.84 ± 0.21 | 0.66 | ||||
| Urea, mg/dL | 31.7 ± 13.0 | 78 | 32.0 ± 15.0 | 31.0 ± 5.9 | 22 | 0.58 | ||
| Glomerular filtration rate, % | 91.7 ± 26.0 | 92.9 ± 27.0 | 88.9 ± 23.7 | 0.26 | ||||
| NT-proBNP, ng/L | 593.3 ± 1003.2 | 78 | 541.2 ± 961.6 | 725.9 ± 1114.6 | 22 | 0.45 | ||
| Diffusion capacity of the lung for carbon monoxide, % | 61.1 ± 17.6 | 78 | 58.1 ± 18.9 | 55 | 68.3 ± 11.1 | 0.019 * | ||
| 6 min walking distance, m | 460.4 ± 112.8 | 78 | 449.8 ± 121.1 | 55 | 485.7 ± 87.2 | 0.185 | ||
| Echocardiography | ||||||||
| Right atrial area, cm2 | 17.2 ± 7.6 | 78 | 16.1 ± 6.3 | 20.1 ± 9.8 | 221 | 0.154 | ||
| Right ventricular area, cm2 | 19.0 ± 7.0 | 72 | 17.9 ± 6.0 | 22.8 ± 9.1 | 16 | 0.022 * | ||
| Systolic pulmonary arterial pressure, mmHg | 53.9 ± 27.0 | 48.0 ± 21.7 | 68.2 ± 34.1 | 0.001 * | ||||
| Tricuspid annular plane systolic excursion, cm | 3.30 ± 3.90 | 77 | 2.35 ± 0.49 | 2.02 ± 0.36 | 21 | 0.014 * | ||
| Right heart catheter at diagnosis | ||||||||
| Mean pulmonary arterial pressure, mmHg | 50.7 ± 14.8 | 74 | 48.4 ± 14.4 | 53 | 56.7 ± 14.6 | 21 | 0.029 * | |
| Cardiac output, L/min | 4.3 ± 1.2 | 55 | 4.6 ± 1.2 | 39 | 3.6 ± 0.9 | 16 | 0.003 * | |
| Pulmonary arterial wedge pressure, mmHg | 9.2 ± 3.4 | 66 | 9.3 ± 3.5 | 50 | 8.8 ± 3.1 | 16 | 0.65 | |
| Pulmonary vascular resistance, WU | 10.6 ± 7.1 | 58 | 9.1 ± 7.0 | 41 | 14.7 ± 6.3 | 17 | <0.001 * | |
| Cardiac index, L/min/m2 | 2.3 ± 0.6 | 50 | 2.5 ± 0.6 | 39 | 2.0 ± 0.4 | 16 | 0.005 * | |
| PAH medication + | ||||||||
| Calcium channel blocker | 1 (1.3) | 1 (1.8) | 0 (0) | |||||
| Mono therapy | 12 (15.2) | 11 (19.6) | 1 (4.4) | |||||
| Double therapy | 39 (49.4) | 28 (50.0) | 11 (47.8) | |||||
| Triple therapy | 27 (34.2) | 16 (28.6) | 11 (47.8) | |||||
| 1/ΔCT Relative BMPR2 Expression | |||
|---|---|---|---|
| Pearson Correlation Coefficient | p-Value | n # | |
| Laboratory | |||
| Hematocrit | −0.242 | 0.031 * | |
| Urea, mg/dL | −0.242 | 0.033 * | 78 |
| Glomerular filtration rate, % | 0.329 | 0.003 * | |
| Creatinine, mg/dL | 0.092 | 0.420 | |
| NT-proBNP, ng/L | −0.204 | 0.073 | 78 |
| Right heart catheterization | |||
| Systolic pulmonary artery pressure (PAP), mmHg | −0.269 | 0.031 * | 65 |
| Diastolic PAP, mmHg | −0.118 | 0.349 | 65 |
| Mean PAP, mmHg | −0.192 | 0.102 | 74 |
| Pulmonary artery wedge pressure, mmHg | −0.047 | 0.710 | 66 |
| Cardiac output, L/min | 0.216 | 0.113 | 55 |
| Pulmonary vascular resistance, WU | −0.278 | 0.035 * | 58 |
| Cardiac index, L/min/m2 | 0.285 | 0.035 * | 55 |
| Transthoracic echocardiography | |||
| Right ventricular wall thickness | −0.282 | 0.014 * | 76 |
| TAPSE, cm | −0.151 | 0.191 | 77 |
| Right atrial area, cm2 | −0.162 | 0.156 | 78 |
| Right ventricular area, cm2 | −0.169 | 0.157 | 72 |
| Systolic PAP, mmHg | −0.271 | 0.016 * | |
| Exercise status | |||
| 6 min walking distance, m | 0.344 | 0.002 * | 78 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Theobald, V.; Benjamin, N.; Seyfarth, H.-J.; Halank, M.; Schneider, M.A.; Richtmann, S.; Hinderhofer, K.; Xanthouli, P.; Egenlauf, B.; Seeger, R.; et al. Reduction of BMPR2 mRNA Expression in Peripheral Blood of Pulmonary Arterial Hypertension Patients: A Marker for Disease Severity? Genes 2022, 13, 759. https://doi.org/10.3390/genes13050759
Theobald V, Benjamin N, Seyfarth H-J, Halank M, Schneider MA, Richtmann S, Hinderhofer K, Xanthouli P, Egenlauf B, Seeger R, et al. Reduction of BMPR2 mRNA Expression in Peripheral Blood of Pulmonary Arterial Hypertension Patients: A Marker for Disease Severity? Genes. 2022; 13(5):759. https://doi.org/10.3390/genes13050759
Chicago/Turabian StyleTheobald, Vivienne, Nicola Benjamin, Hans-Jürgen Seyfarth, Michael Halank, Marc A. Schneider, Sarah Richtmann, Katrin Hinderhofer, Panagiota Xanthouli, Benjamin Egenlauf, Rebekka Seeger, and et al. 2022. "Reduction of BMPR2 mRNA Expression in Peripheral Blood of Pulmonary Arterial Hypertension Patients: A Marker for Disease Severity?" Genes 13, no. 5: 759. https://doi.org/10.3390/genes13050759
APA StyleTheobald, V., Benjamin, N., Seyfarth, H.-J., Halank, M., Schneider, M. A., Richtmann, S., Hinderhofer, K., Xanthouli, P., Egenlauf, B., Seeger, R., Hoeper, M. M., Jonigk, D., Grünig, E., & Eichstaedt, C. A. (2022). Reduction of BMPR2 mRNA Expression in Peripheral Blood of Pulmonary Arterial Hypertension Patients: A Marker for Disease Severity? Genes, 13(5), 759. https://doi.org/10.3390/genes13050759