
Expression Characteristics in Roots, Phloem, Leaves, Flowers and Fruits of Apple circRNA




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Materials
2.2. Genome-Wide circRNAs Sequencing
2.3. Identification and Differential Expression Profile of circRNAs in Apple Tissues
2.4. Functional Annotation Analysis of Parental Genes of circRNAs
2.5. Prediction of miRNA Target Sites in circRNAs
2.6. Quantitative Real-Time PCR Validation
3. Results
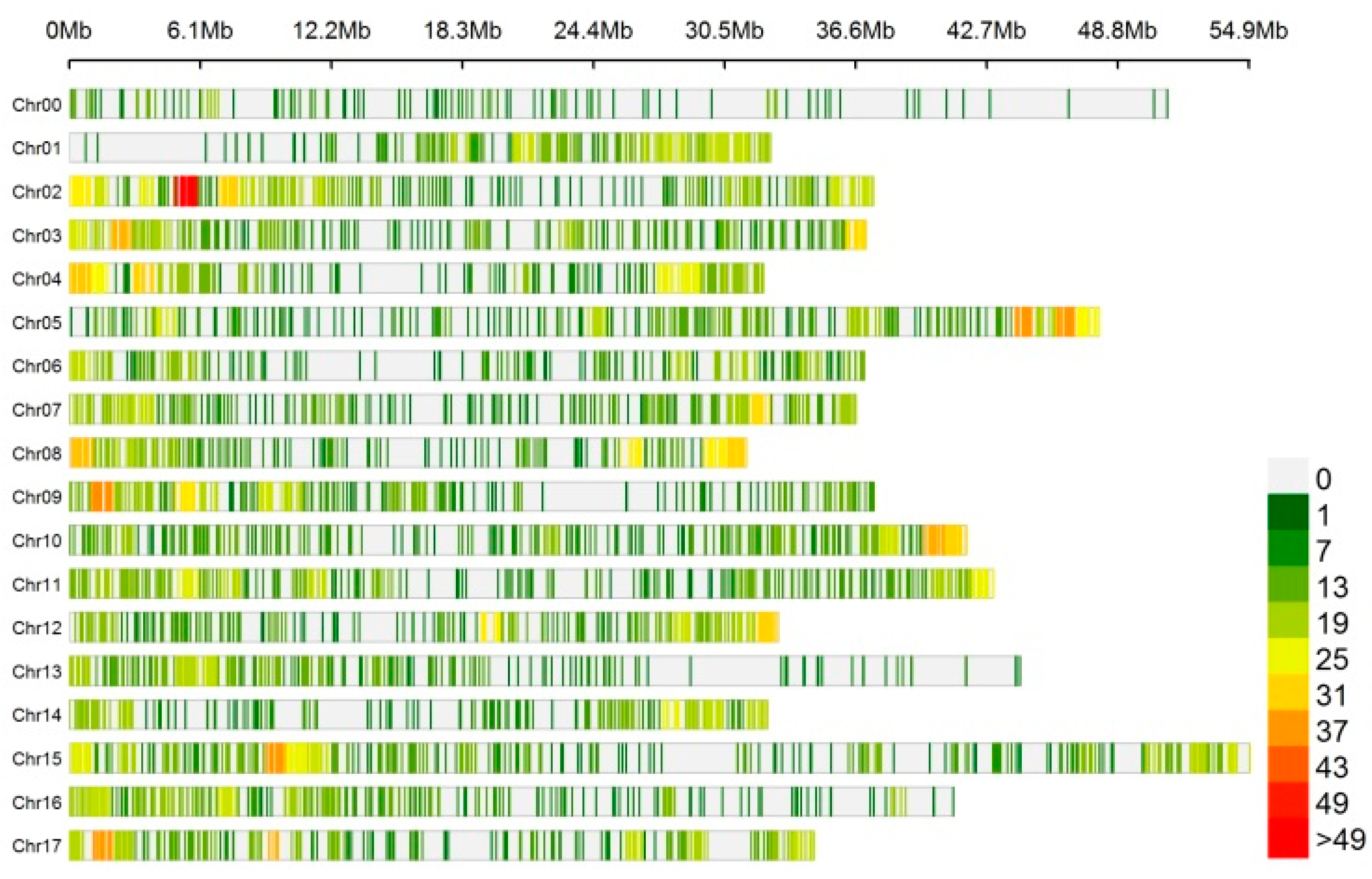
3.1. Identification and Characteristics of circRNAs in ‘Gala’ Apple
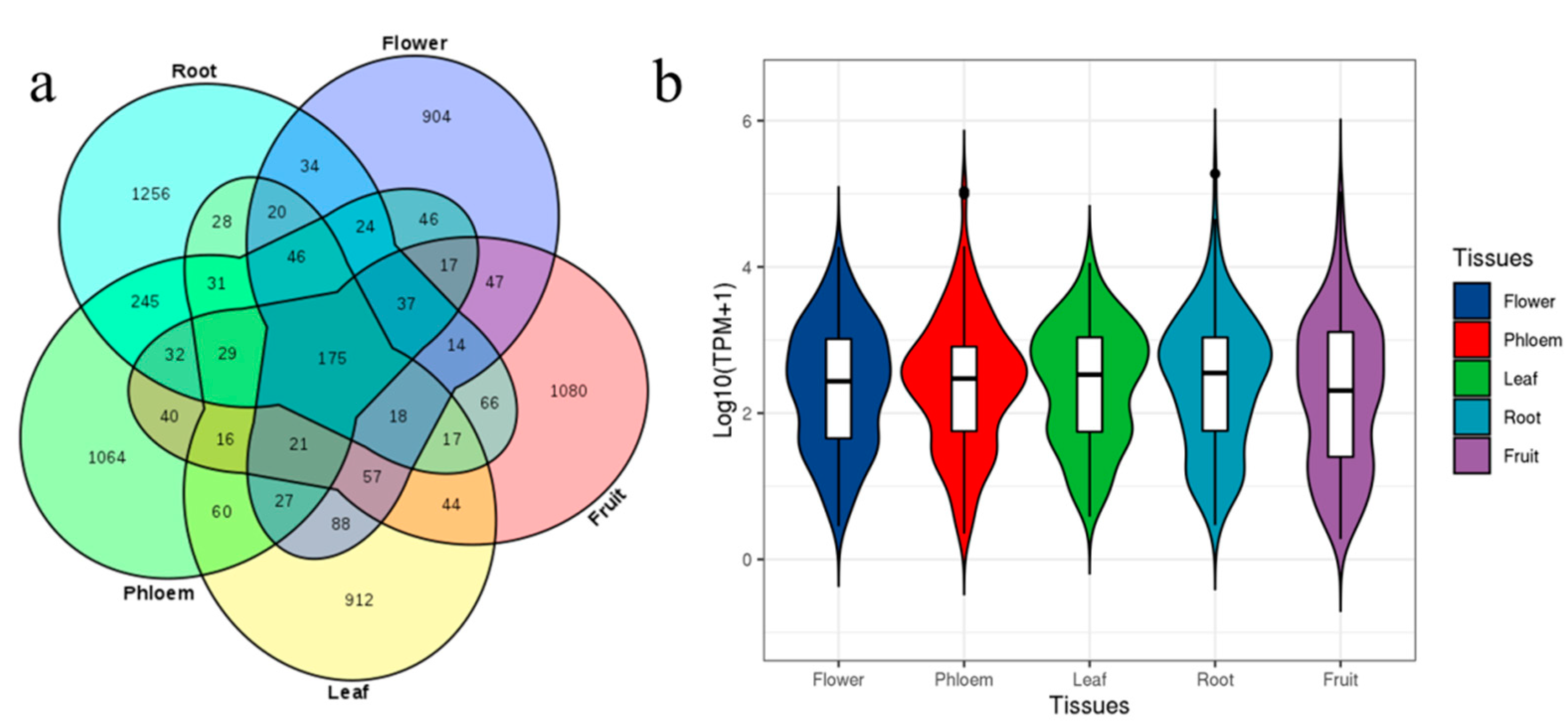
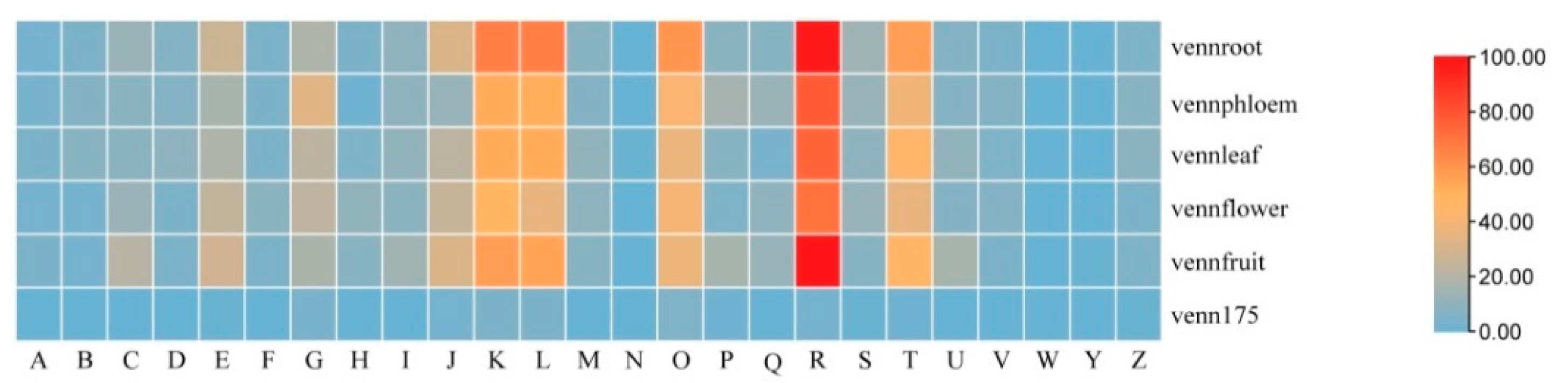
3.2. Different Expression in Tissues of ‘Gala’ Apple
3.3. Functional Annotation of Host-Genes of circRNAs
3.4. CircRNA–miRNA Interaction Network in Apple
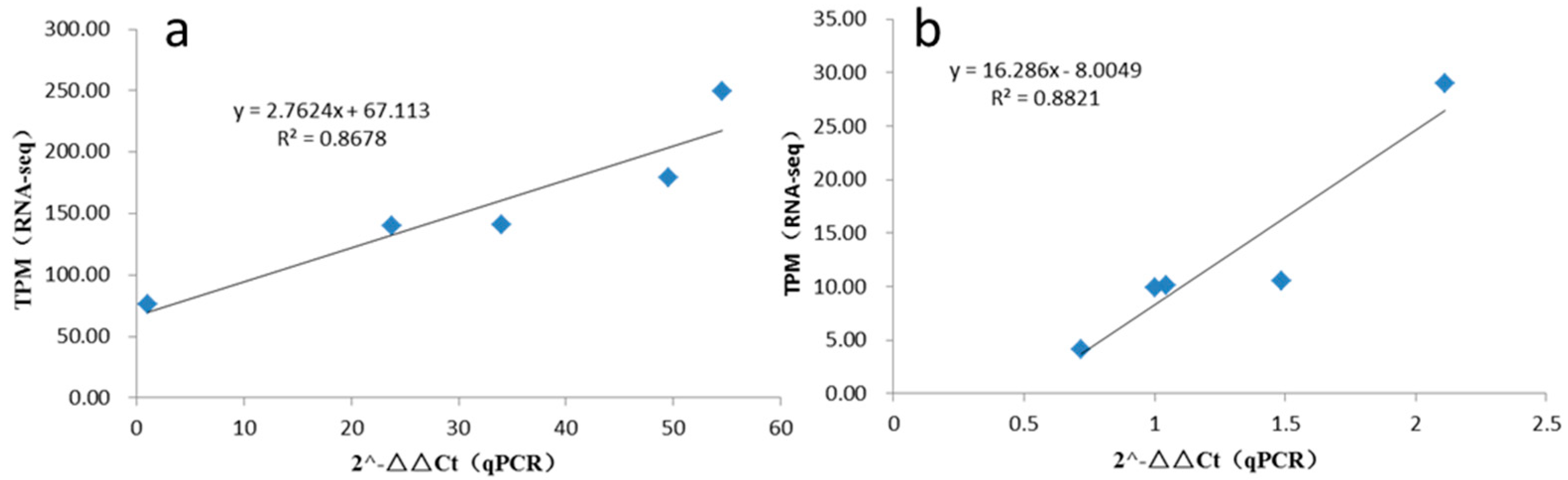
3.5. qRT-PCR Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanger, H.L.; Klotz, G.; Riesner, D.; Gross, H.J.; Kleinschmidt, A.K. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc. Natl. Acad. Sci. USA 1976, 73, 3852–3856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. CircRNA biogenesis competes with pre-mRNA splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, L.; Salzman, J. Detecting circular RNAs: Bioinformatic and experimental challenges. Nat. Rev. Genet. 2016, 17, 679–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.L.; Bao, Y.; Yee, M.C.; Barrett, S.P.; Hogan, G.J.; Olsen, M.N.; Dinneny, J.R.; Brown, P.O.; Salzman, J.; Thomas, P. Circular RNA is expressed across the eukaryotic tree of life. PLoS ONE 2014, 9, e90859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.; Luo, M.; Wang, L.; Song, S.R.; Zhao, L.P.; Xu, W.P.; Zhang, C.X.; Wang, S.P.; Ma, C. The research advance of plant circular RNA. Acta Hortic. Sin. 2019, 46, 171–181. [Google Scholar] [CrossRef]
- Lasda, E.; Parker, R. Circular RNAs: Diversity of form and function. RNA 2014, 20, 1829–1842. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.O.; Wang, H.B.; Zhang, Y.; Chen, L.; Yang, L. Complementary sequence-mediated exon circularization. Cell 2014, 159, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.T.; Cui, L.L.; Zhou, Y.; Zhu, C.R.; Fan, D.L.; Gong, H.; Zhao, Q.; Zhou, C.C.; Zhao, Y.; Lu, D.F.; et al. Transcriptome-wide investigation of circular RNAs in rice. RNA 2015, 21, 2076–2087. [Google Scholar] [CrossRef] [Green Version]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef]
- Lee, S.M.; Kong, H.G.; Ryu, C.M. Are circular RNAs new kids on the block? Trends Plant Sci. 2017, 22, 357–360. [Google Scholar] [CrossRef]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhang, X.O.; Chen, T.; Xiang, J.F.; Yin, Q.F.; Xing, Y.H.; Zhu, S.; Yang, L.; Chen, L.L. Circular intronic long noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salzman, J.; Chen, R.E.; Olsen, M.N.; Wang, P.L.; Brown, P.O. Cell-type specific features of circular RNA expression. PLoS Genet. 2013, 9, e1003777. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [Green Version]
- Ye, C.Y.; Chen, L.; Liu, C.; Zhu, Q.H.; Fan, L. Widespread noncoding circular RNAs in plants. New Phytol. 2015, 208, 88–95. [Google Scholar] [CrossRef]
- Li, Z.; Wang, S.P.; Cheng, J.P.; Su, C.B.; Zhong, S.X.; Liu, Q.; Fang, Y.D.; Yu, Y.; Lv, H.; Zheng, Y.; et al. Intron lariat RNA inhibits microRNA biogenesis by sequestering the dicing complex in Arabidopsis. PLoS Genet. 2016, 12, e1006422. [Google Scholar] [CrossRef]
- Chen, G.; Cui, J.W.; Wang, L.; Zhu, Y.F.; Lu, Z.G.; Jin, B. Genome-wide identification of circular RNAs in Arabidopsis thaliana. Front. Plant Sci. 2017, 8, 1678. [Google Scholar] [CrossRef]
- Wang, Y.X.; Yang, M.; Wei, S.M.; Qin, F.J.; Zhao, H.J.; Suo, B. Identification of circular RNAs and their targets in leaves of Triticum aestivum L. under dehydration stress. Front. Plant Sci. 2017, 7, 2024. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.P.; Liu, Y.F.; Li, D.W.; Li, L.; Zhang, Q.; Wang, S.B.; Huang, H.W. Identification of circular RNAs in kiwifruit and their species-specific response to bacterial canker pathogen invasion. Front. Plant Sci. 2017, 8, 413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, T.; Sun, X.Q.; Liu, Y.X.; Li, H.; Deng, G.B.; Lin, H.H.; Wang, S.H. Heat stress alters genome-wide profiles of circular RNAs in Arabidopsis. Plant Mol. Biol. 2018, 96, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.F.; Zhou, J.J.; Hu, C.G.; Zhang, J.Z. Transcriptome-wide identification and functional prediction of novel and flowering-related circular RNAs from trifoliate orange(Poncirus trifoliata L. Raf.). Planta 2018, 247, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Velasco, R.; Zharkikh, A.; Affourtit, J.; Dhingra, A.; Cestaro, A.; Kalyanaraman, A.; Fontana, P.; Bhatnagar, S.K.; Troggio, M.; Pruss, D. The genome of the domesticated apple (Malus × domestica Borkh.). Nat. Genet. 2010, 42, 833–839. [Google Scholar] [CrossRef]
- Li, X.W.; Ling, K.; Zhang, J.; Xie, Y.P.; Wang, L.P.; Yan, Y.; Wang, N.; Xu, J.D.; Li, C.Y.; Wang, W.; et al. Improved hybrid de novo genome assembly of domesticated apple (Malus × domestica). GigaScience 2016, 5, 35. [Google Scholar] [CrossRef] [Green Version]
- Daccord, N.; Celton, J.M.; Linsmith, G.; Becker, C.; Choisne, N.; Schijlen, E.; Henri, V.D.G.; Bianco, L.; Micheletti, D.; Velasco, R. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 2017, 49, 1099–1106. [Google Scholar] [CrossRef]
- Zhang, L.Y.; Hu, J.; Han, X.L.; Li, J.J.; Gao, Y.; Richards, C.M.; Zhang, C.X.; Tian, Y.; Liu, G.M.; Gul, H.; et al. A high-quality apple genome assembly reveals the association of a retrotransposon and red fruit colour. Nat. Commun. 2019, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.P.; Jiao, C.; Schwaninger, H.; Chao, C.T.; Ma, Y.M.; Duan, N.B.; Khan, A.; Ban, S.; Xu, K.; Cheng, L.L.; et al. Phased diploid genome assemblies and pan-genomes provide insights into the genetic history of apple domestication. Nat. Genet. 2020, 52, 1423–1432. [Google Scholar] [CrossRef]
- Darbani, B.; Noeparvar, S.; Borg, S. Identification of circular RNAs from the parental genes involved in multiple aspects of cellular metabolism in barley. Front. Plant Sci. 2016, 7, 776. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.F.; Zhang, L.; Chen, G.; Shi, T.L. Identifying and characterizing the circular RNAs during the lifespan of Arabidopsis leaves. Front. Plant Sci. 2017, 8, 1278. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Wu, L.; Qi, H.R.; Xu, M. LncRNA/circRNA-miRNA-mRNA networks regulate the development of root and shoot meristems of Populus. Ind. Crops Prod. 2019, 133, 333–347. [Google Scholar] [CrossRef]
- Wang, J.X.; Lin, J.; Wang, H.; Li, X.G.; Yang, Q.S.; Li, H.; Chang, Y.H.; Min, Y.Z. Identification and characterization of circRNAs in Pyrus betulifolia Bunge under drought stress. PLoS ONE 2018, 13, e0200692. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.Y.; Feng, J.; Lei, L.J.; Hu, J.; Xia, L.J.; Wang, J.; Xiang, Y.; Liu, L.J.; Zhong, S.; Han, L.; et al. Comprehensive characterization of tissue-specific circular RNAs in the human and mouse genomes. Brief Bioinform. 2017, 18, 984–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Zhang, P.; Fan, Y.; Lu, Q.; Li, Q.; Yan, J.B.; Muehlbauer, G.J.; Schnable, P.S.; Dai, M.Q.; Li, L. Circular RNAs mediated by transposons are associated with transcriptomic and phenotypic variation in maize. New Phytol. 2018, 217, 1292–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, W.W.; Li, H.M.; Qing, X.R.; Huang, D.H.; Li, H.G. Identification and characterization of human testis derived circular RNAs and their existence in seminal plasma. Sci. Rep. 2016, 6, 39080. [Google Scholar] [CrossRef] [Green Version]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Zuo, J.H.; Wang, Q.; Zhu, B.Z.; Luo, Y.B.; Gao, L.P. Deciphering the roles of circRNAs on chilling injury in tomato. Biochem. Biophys. Res. Commun. 2016, 479, 132–138. [Google Scholar] [CrossRef]
- Yin, J.L.; Liu, M.Y.; Ma, D.F.; Wu, J.W.; Li, S.L.; Zhu, Y.X.; Han, B. Identification of circular RNAs and their targets during tomato fruit ripening. Postharvest Biol. Technol. 2018, 136, 90–98. [Google Scholar] [CrossRef]
- Subha, D.; Shira, C.; Kumar, G.S.; Tzahi, A. Tuning of SlARF10A dosage by sly-miR160a is critical for auxin-mediated compound leaf and flower development. Plant J. 2018, 96, 855–868. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.; Mao, J.P.; Tahir, M.M.; Wang, H.; Wei, Y.H.; Zhao, C.D.; Li, K.; Ma, D.D.; Zhao, C.P.; Zhang, D. Mdm-miR160 Participates in Auxin-Induced Adventitious Root formation of apple rootstock. Sci. Hortic. 2020, 270, 109442. [Google Scholar] [CrossRef]
- Luo, Y.L.; Li, Y.X.; Zhang, J.; Zhang, J.; Yao, Y.C. Cloning and functional assay of McmiR160 regulated leaf coloration in Malus spp. J. Beijing Univ. Agric. 2019, 34, 14–19. [Google Scholar] [CrossRef]
- Subramanina, S.; Fu, Y.; Sunkar, R.; Barbazuk, W.B.; Zhu, J.K.; Yu, O. Novel and nodulation-regulated microRNAs in soybean roots. BMC Genom. 2008, 9, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuda, N.; Isono, T.; Sato, M.H. Arabidopsis CAMTA family proteins enhance V-PPase expression in pollen. Plant Cell Physiol. 2003, 44, 975–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, Y.; Nave, R.; Boyce, J.M.; Nachmias, D.; Knight, M.R.; Fromm, H. Calmodulin-binding transcription activator (CAMTA) 3 mediates biotic defense responses in Arabidopsis. FEBS Lett. 2008, 582, 943–948. [Google Scholar] [CrossRef] [Green Version]
- Doherty, C.J.; Van Buskirk, H.A.; Myers, S.J.; Thomashow, M.F. Roles for Arabidopsis CAMTA transcription factors in cold-regulated gene expression and freezing tolerance. Plant Cell 2009, 21, 972–984. [Google Scholar] [CrossRef] [Green Version]
- Koo, S.C.; Choi, M.S.; Chun, H.J.; Dong, B.S.; Park, B.S.; Kim, Y.H.; Park, H.M.; Seo, H.S.; Song, J.T.; Kang, K.Y. The calmodulin-binding transcription factor OsCBT suppresses defense responses to pathogens in rice. Mol. Cells 2009, 27, 563–570. [Google Scholar] [CrossRef]
- Laluk, K.; Prasad, K.V.S.K.; Savchenko, T.; Celesnik, H.; Dehesh, K.; Levy, M.; Mitchell-Olds, T.; Reddy, A.S.N. The calmodulin-binding transcription factor SIGNAL RESPONSIVE1 is a novel regulator of glucosinolate metabolism and herbivory tolerance in Arabidopsis. Plant Cell Physiol. 2012, 53, 2008–2015. [Google Scholar] [CrossRef]
- Pandey, N.; Ranjan, A.; Pant, P.; Tripathi, R.K.; Ateek, F.; Pandey, H.P.; Patre, U.V.; Sawant, S.V. CAMTA 1 regulates drought responses in Arabidopsis thaliana. BMC Genom. 2013, 14, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benn, G.; Wang, C.Q.; Hicks, D.R.; Stein, J. A key general stress response motif is regulated non-uniformly by CAMTA transcription factors. Plant J. 2014, 80, 82–92. [Google Scholar] [CrossRef]
- Li, X.H.; Huang, L.; Zhang, Y.F.; Ouyang, Z.G.; Hong, Y.B.; Zhang, H.J.; Li, D.Y.; Song, F.M. Tomato SR/CAMTA transcription factors SlSR1 and SlSR3L negatively regulate disease resistance response and SlSR1L positively modulates drought stress tolerance. BMC Plant Biol. 2014, 14, 286. [Google Scholar] [CrossRef]
- Kim, S.M.; Suh, J.P.; Lee, C.K.; Lee, J.H.; Kim, Y.G.; Jena, K.K. QTL mapping and development of candidate gene-derived DNA markers associated with seedling cold tolerance in rice (Oryza sativa L.). Mol. Genet. Genom. 2014, 289, 333–343. [Google Scholar] [CrossRef]
- Prasad, K.V.S.K.; Abdel-Hameed, A.A.E.; Xing, D.; Reddy, A.S.N. Global gene expression analysis using RNA-seq uncovered a new role for SR1/CAMTA3 transcription factor in salt stress. Sci. Rep. 2016, 6, 27021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.G.; Liu, Y.H.; Liu, Z.C.; Kong, D.Y.; Duan, M.; Luo, L.J. Genome-wide identification and analysis of drought-responsive microRNAs in Oryza sativa. J. Exp. Bot. 2010, 61, 4157–4168. [Google Scholar] [CrossRef] [PubMed]
- Capitão, C.; Paiva, J.A.P.; Santos, D.M.; Fevereiro, P. In Medicago truncatula, water deficit modulates the transcript accumulation of components of small RNA pathways. BMC Plant Biol. 2011, 11, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Cui, X.; Meng, Z.L.; Huang, X.H.; Xie, Q.; Wu, H.; Jin, H.L.; Zhang, D.B.; Liang, W.Q. Transcriptional regulation of Arabidopsis MIR168a and ARGONAUTE1 homeostasis in abscisic acid and abiotic stress responses. Plant Physiol. 2012, 158, 1279–1292. [Google Scholar] [CrossRef] [Green Version]
- Li, D.T.; Wang, L.W.; Liu, X.; Cui, D.Z.; Chen, T.T.; Zhang, H.; Jiang, C.; Xu, C.Y.; Li, P.; Li, S.; et al. Deep sequencing of maize small RNAs reveals a diverse set of microRNA in dry and imbibed seeds. PLoS ONE 2013, 8, e55107. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Gao, Y.; Sun, S.; Li, L.; Wang, K. Expression Characteristics in Roots, Phloem, Leaves, Flowers and Fruits of Apple circRNA. Genes 2022, 13, 712. https://doi.org/10.3390/genes13040712
Wang D, Gao Y, Sun S, Li L, Wang K. Expression Characteristics in Roots, Phloem, Leaves, Flowers and Fruits of Apple circRNA. Genes. 2022; 13(4):712. https://doi.org/10.3390/genes13040712
Chicago/Turabian StyleWang, Dajiang, Yuan Gao, Simiao Sun, Lianwen Li, and Kun Wang. 2022. "Expression Characteristics in Roots, Phloem, Leaves, Flowers and Fruits of Apple circRNA" Genes 13, no. 4: 712. https://doi.org/10.3390/genes13040712
APA StyleWang, D., Gao, Y., Sun, S., Li, L., & Wang, K. (2022). Expression Characteristics in Roots, Phloem, Leaves, Flowers and Fruits of Apple circRNA. Genes, 13(4), 712. https://doi.org/10.3390/genes13040712