
Dietary Restriction and Rapamycin Affect Brain Aging in Mice by Attenuating Age-Related DNA Methylation Changes

Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Animals and Library Preparation
2.2. DNA Methylation Data Analysis
2.3. Gene Expression Data Processing
2.4. GO Enrichment Analysis of DR/RALL-Related Genes
3. Results
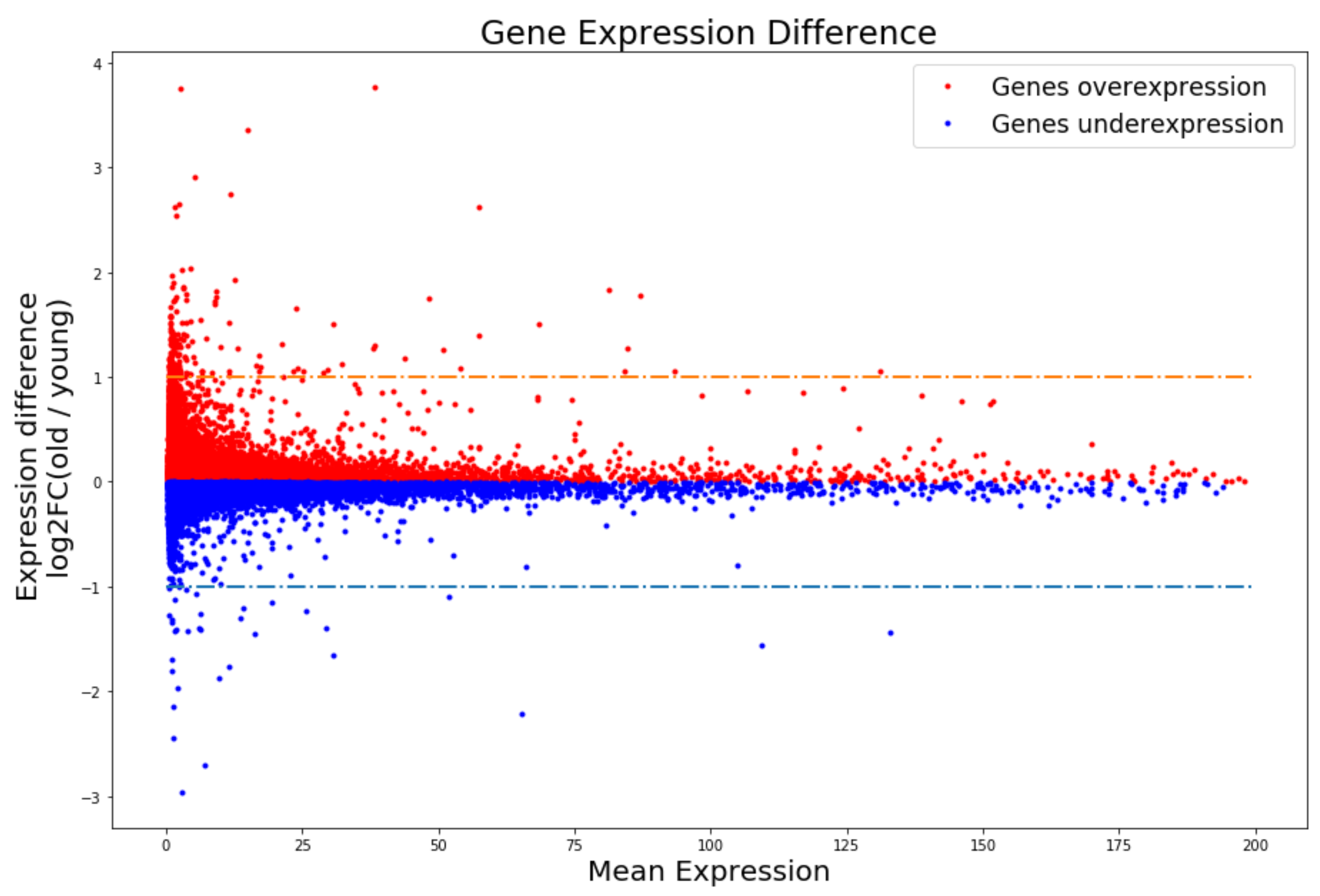
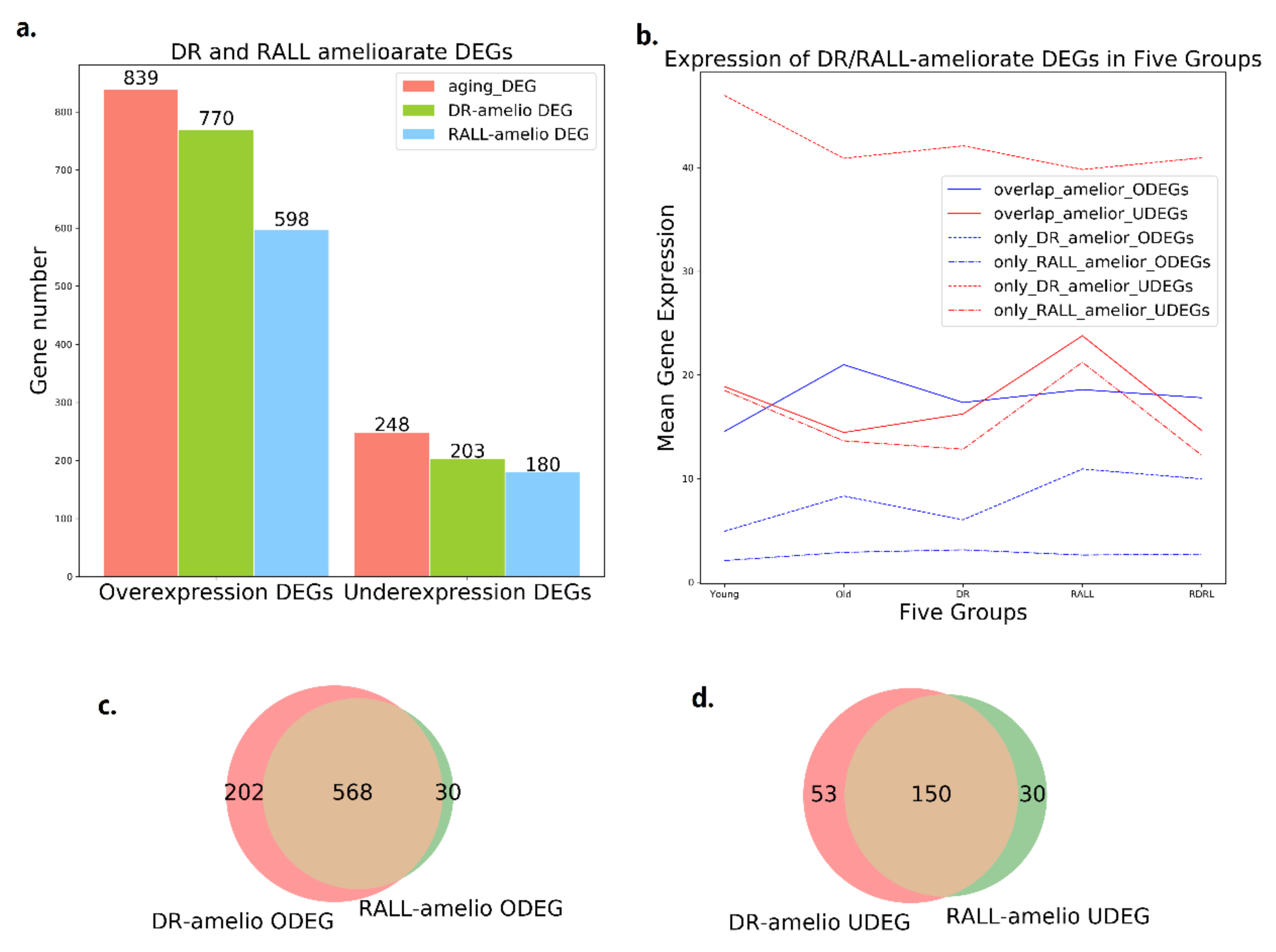
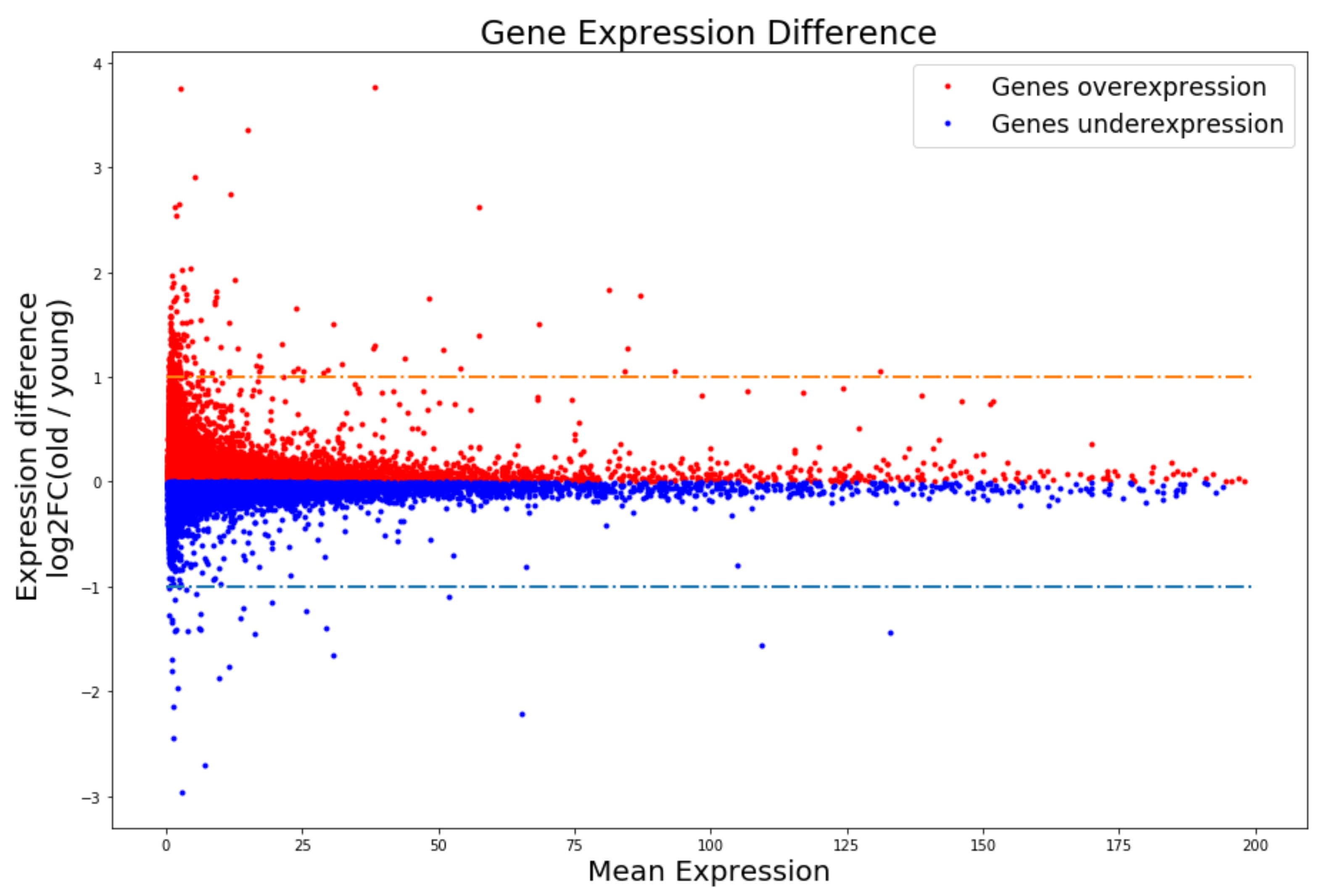
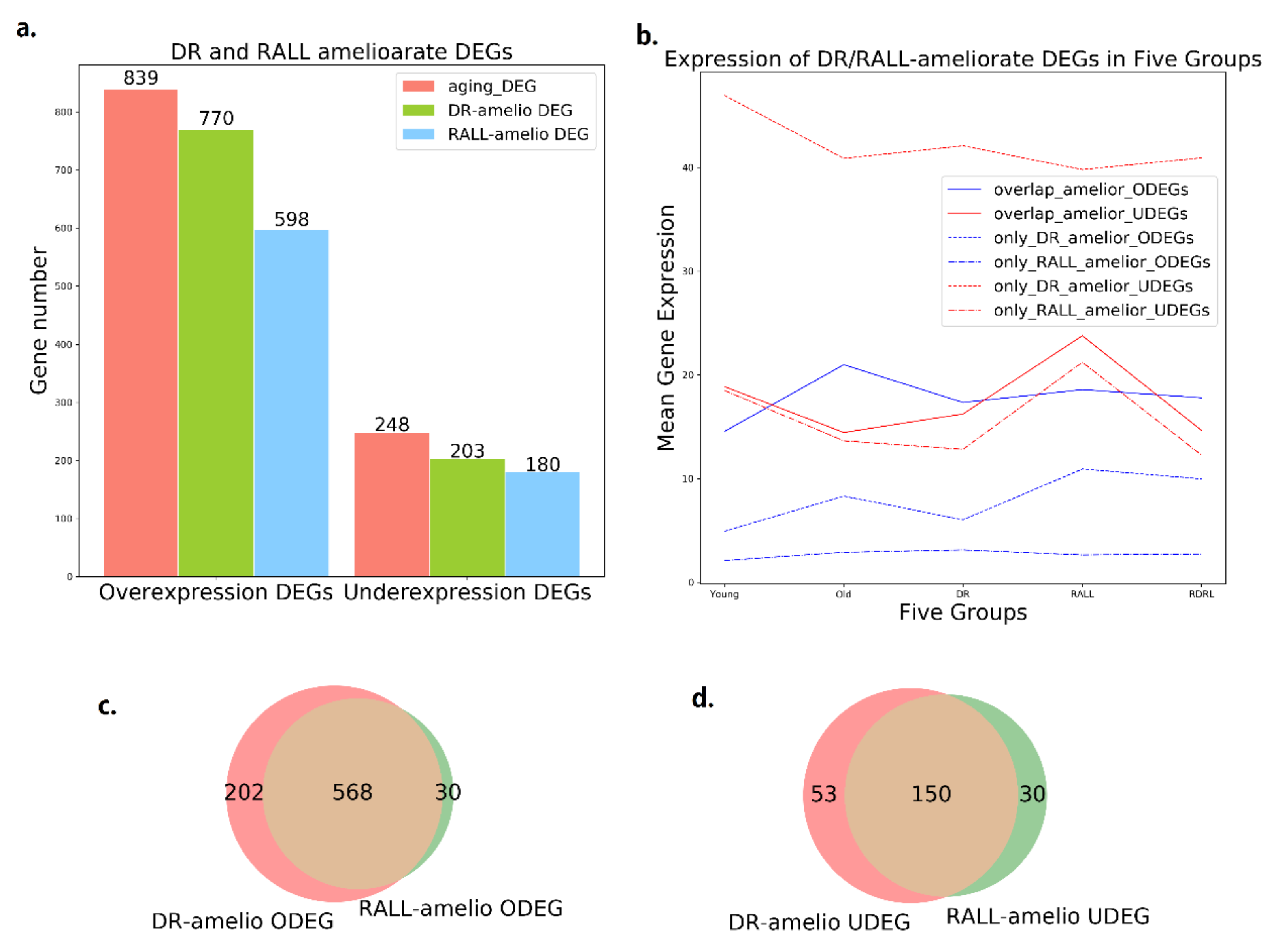
3.1. DR and RALL Can Attenuate Gene Expression Changes Associated with Aging
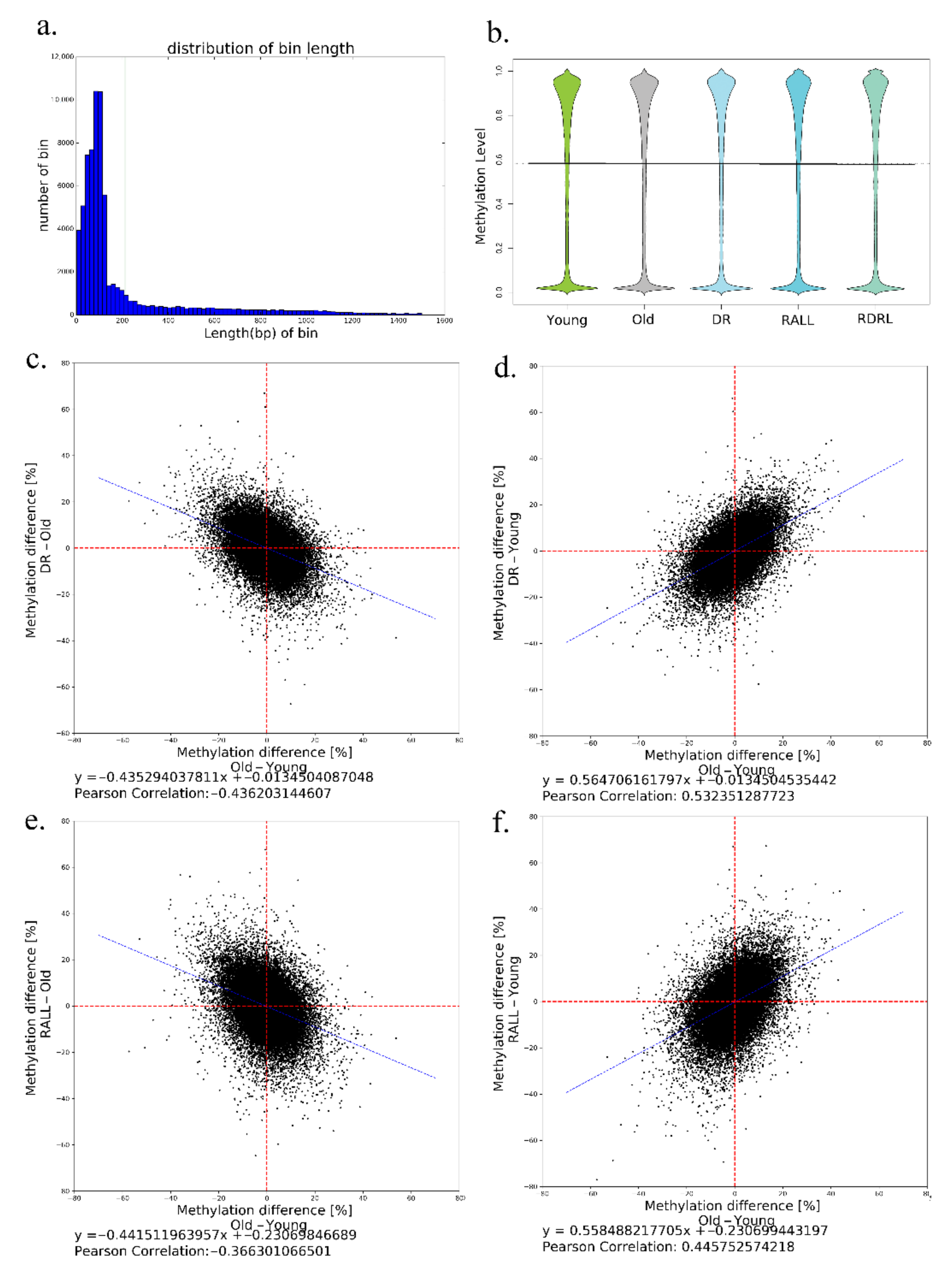
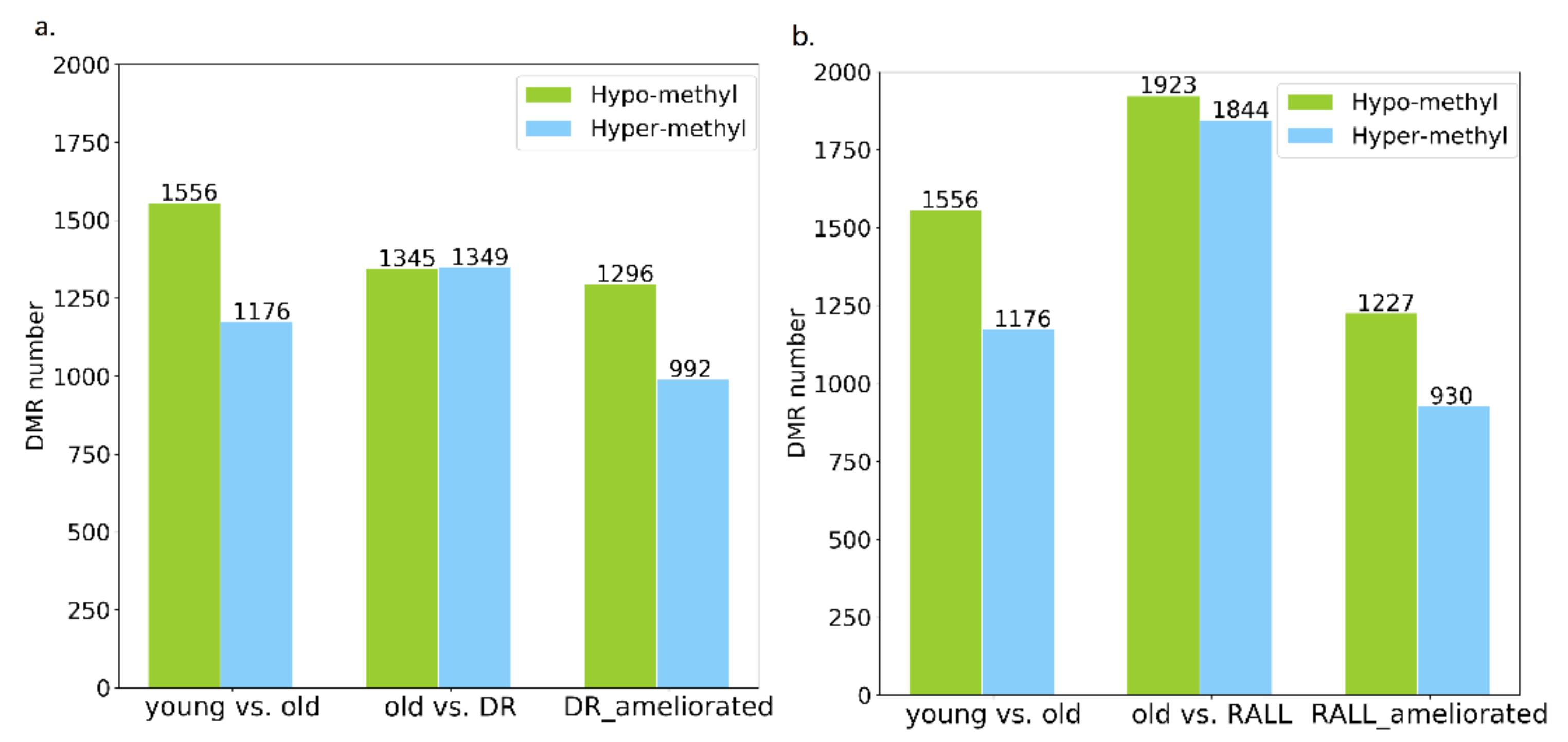
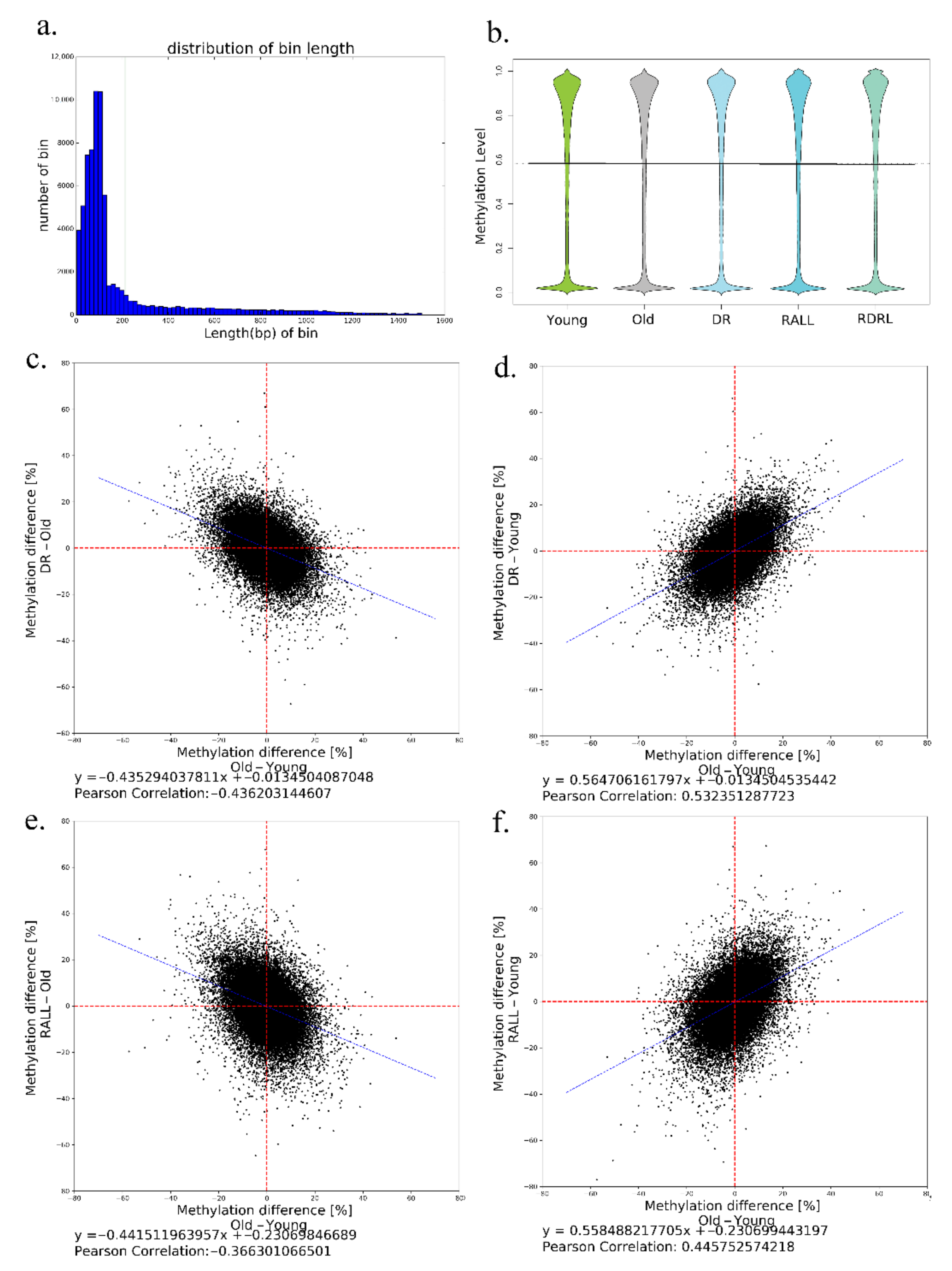
3.2. DR and RALL Can Globally Attenuate Aging-Related Methylation Changes
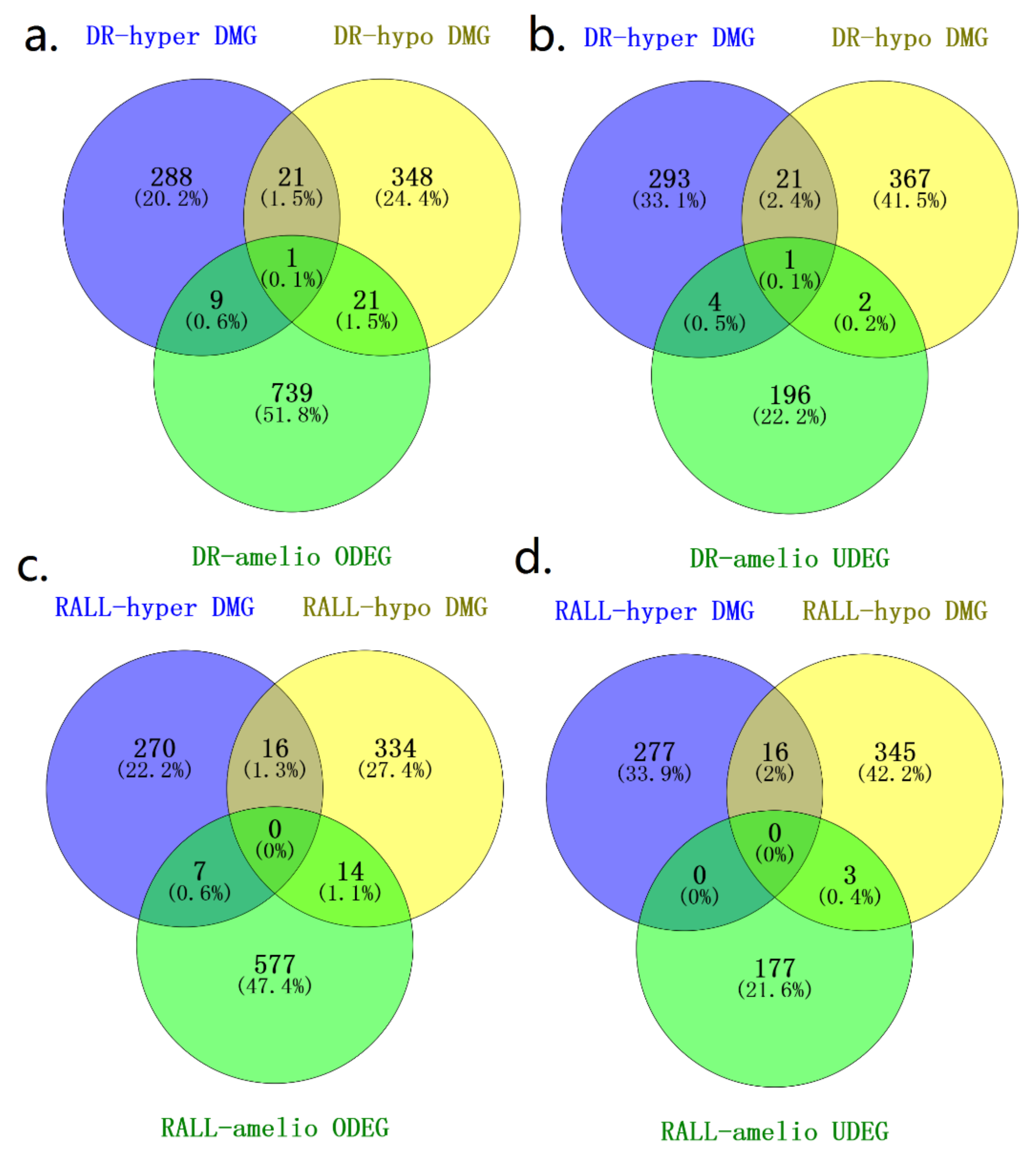
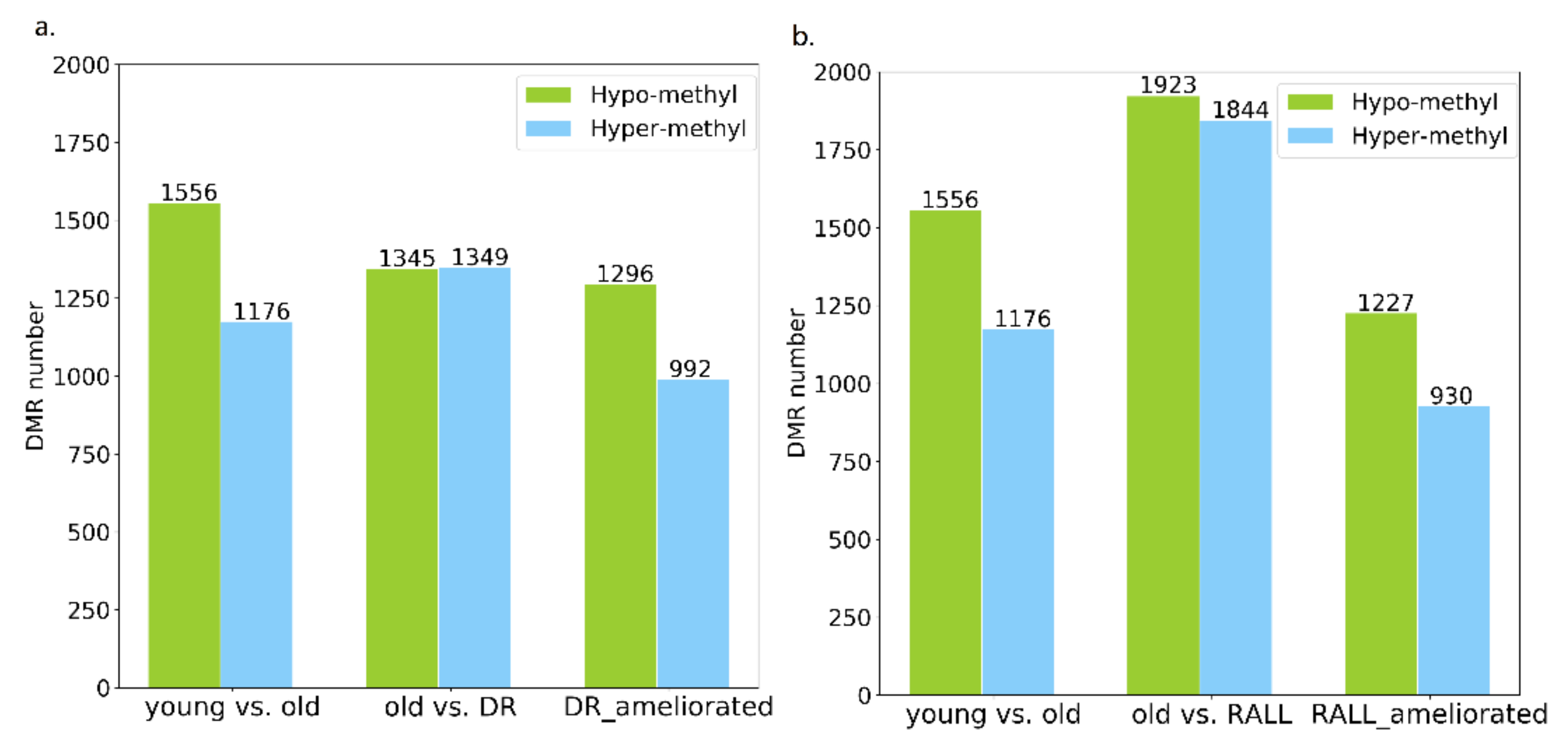
3.3. Aging-Related Methylation Changes Could Be Attenuated by DR and RALL
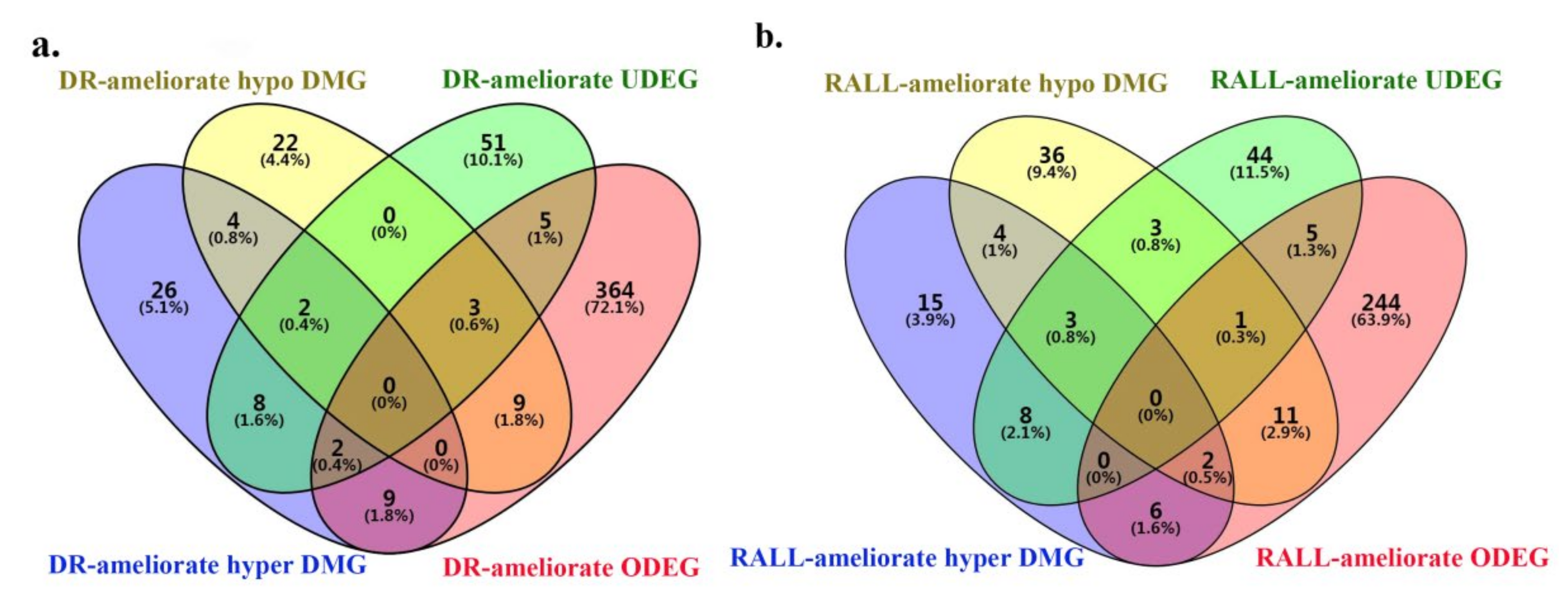
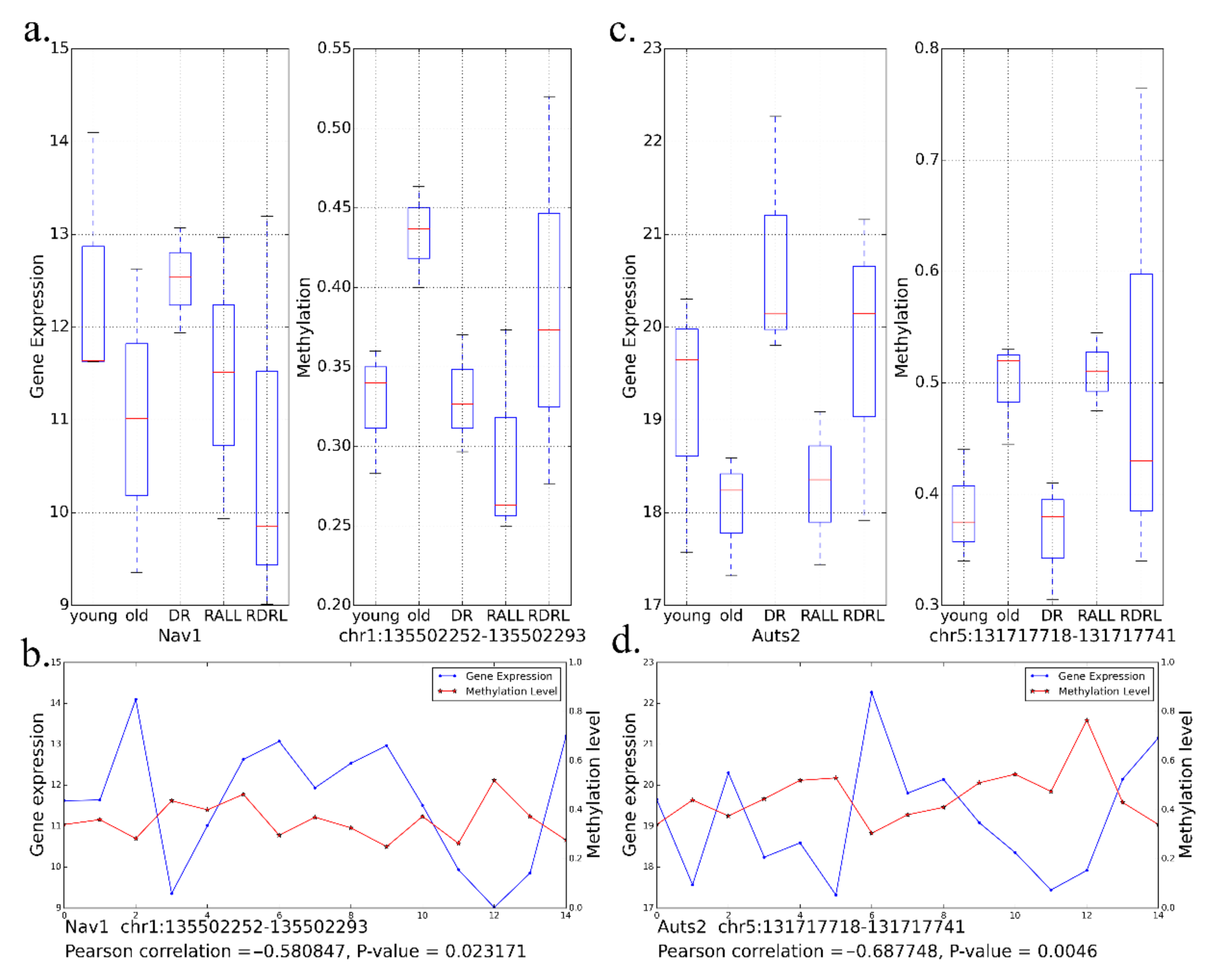
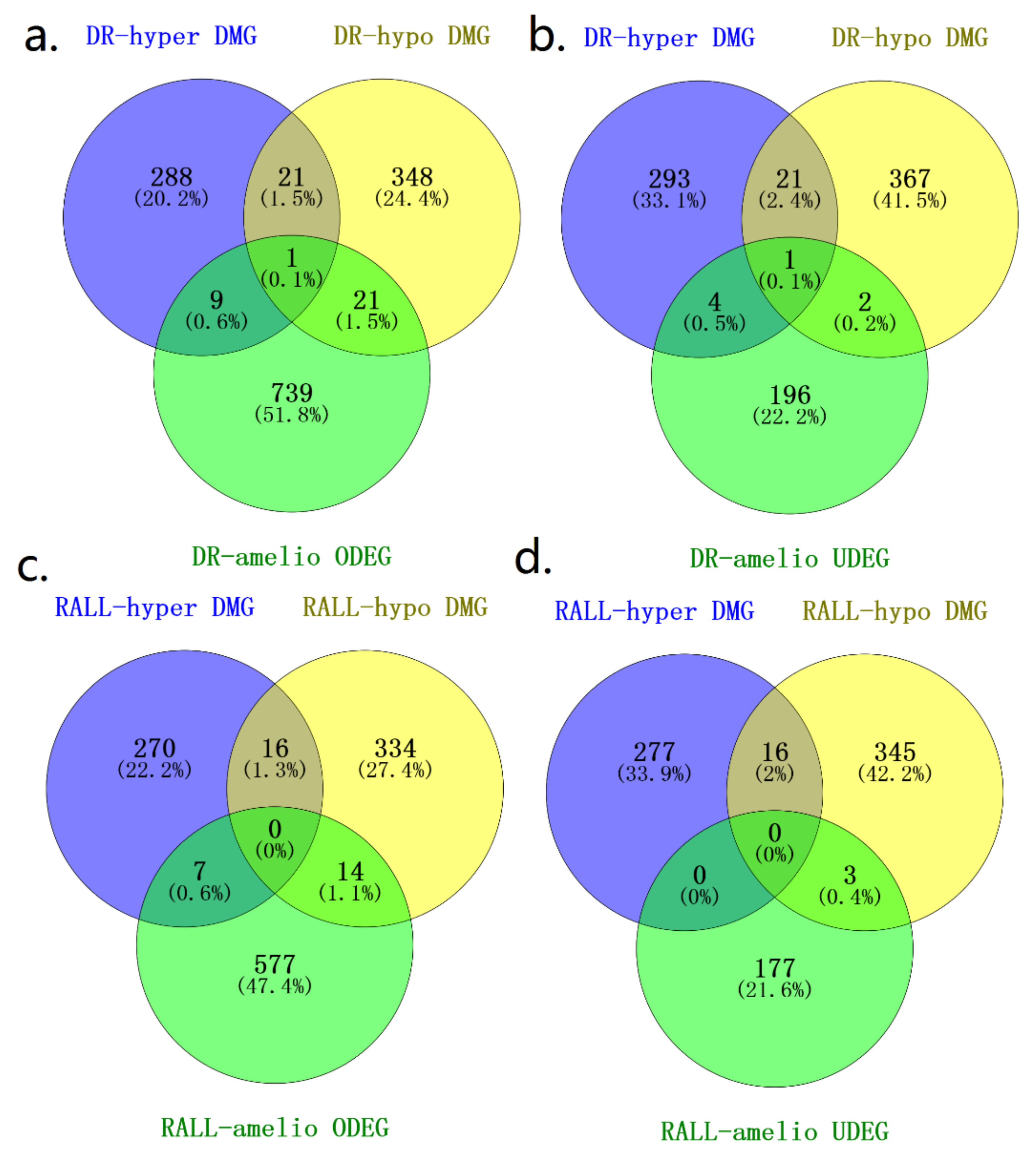
3.4. DR and RALL-Ameliorated Genes Are Associated with Aging-Related Functions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Wang, T.; Tsui, B.; Kreisberg, J.F.; Robertson, N.A.; Gross, A.M.; Yu, M.K.; Carter, H.; Brown-Borg, H.M.; Adams, P.D.; Ideker, T. Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biol. 2017, 18, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, O.; Gronke, S.; Stubbs, T.M.; Ficz, G.; Hendrich, O.; Krueger, F.; Andrews, S.; Zhang, Q.; Wakelam, M.J.; Beyer, A.; et al. Dietary restriction protects from age-associated DNA methylation and induces epigenetic reprogramming of lipid metabolism. Genome Biol. 2017, 18, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Daniel, M.; Tollefsbol, T.O. Epigenetic regulation of caloric restriction in aging. BMC Med. 2011, 9, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, L.N.; Brunet, A. The Aging Epigenome. Mol. Cell 2016, 62, 728–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Nogales-Cadenas, R.; Lin, J.R.; Zhang, W.; Cai, Y.; Vijg, J.; Zhang, Z.D. Systems-level analysis of human aging genes shed new light on mechanisms of aging. Hum. Mol. Genet. 2016, 25, 2934–2947. [Google Scholar] [CrossRef] [Green Version]
- Moskalev, A.A.; Aliper, A.M.; Smit-McBride, Z.; Buzdin, A.; Zhavoronkov, A. Genetics and epigenetics of aging and longevity. Cell Cycle 2014, 13, 1063–1077. [Google Scholar] [CrossRef] [Green Version]
- Benayoun, B.A.; Pollina, E.A.; Brunet, A. Epigenetic regulation of ageing: Linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 2015, 16, 593–610. [Google Scholar] [CrossRef] [Green Version]
- Van Dongen, J.; Ehli, E.A.; Slieker, R.C.; Bartels, M.; Weber, Z.M.; Davies, G.E.; Slagboom, P.E.; Heijmans, B.T.; Boomsma, D.I. Epigenetic variation in monozygotic twins: A genome-wide analysis of DNA methylation in buccal cells. Genes 2014, 5, 347–365. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.; Pfeifer, G.P. Aging and DNA methylation. BMC Biol. 2015, 13, 7. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 16, 3156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibikova, M.; Chudin, E.; Wu, B.; Zhou, L.; Garcia, E.W.; Liu, Y.; Shin, S.; Plaia, T.W.; Auerbach, J.M.; Arking, D.E.; et al. Human embryonic stem cells have a unique epigenetic signature. Genome Res. 2006, 16, 1075–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solovev, I.; Shaposhnikov, M.; Moskalev, A. Multi-omics approaches to human biological age estimation. Mech. Ageing Dev. 2020, 185, 111192. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Sierra, J.V.; Helbing, A.H.B.; Williams, E.G.; Ashbrook, D.G.; Roy, S.; Williams, R.W.; Mozhui, K. Body weight and high-fat diet are associated with epigenetic aging in female members of the BXD murine family. Aging Cell 2020, 19, e13207. [Google Scholar] [CrossRef]
- Galkin, F.; Mamoshina, P.; Aliper, A.; de Magalhaes, J.P.; Gladyshev, V.N.; Zhavoronkov, A. Biohorology and biomarkers of aging: Current state-of-the-art, challenges and opportunities. Ageing Res. Rev. 2020, 60, 101050. [Google Scholar] [CrossRef]
- Widschwendter, M.; Jones, A.; Evans, I.; Reisel, D.; Dillner, J.; Sundström, K.; Steyerberg, E.W.; Vergouwe, Y.; Wegwarth, O.; Rebitschek, F.G.; et al. Epigenome-based cancer risk prediction: Rationale, opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 292–309. [Google Scholar] [CrossRef]
- Yan, H.; He, J.; Guan, Q.; Cai, H.; Zhang, L.; Zheng, W.; Qi, L.; Zhang, S.; Liu, H.; Li, H.; et al. Identifying CpG sites with different differential methylation frequencies in colorectal cancer tissues based on individualized differential methylation analysis. Oncotarget 2017, 8, 47356–47364. [Google Scholar] [CrossRef] [Green Version]
- Gensous, N.; Ravaioli, F.; Pirazzini, C.; Gramignoli, R.; Ellis, E.; Storci, G.; Capri, M.; Strom, S.; Laconi, E.; Franceschi, C.; et al. Aging and Caloric Restriction Modulate the DNA Methylation Profile of the Ribosomal RNA Locus in Human and Rat Liver. Nutrients 2020, 12, 277. [Google Scholar] [CrossRef] [Green Version]
- Farre, P.; Jones, M.J.; Meaney, M.J.; Emberly, E.; Turecki, G.; Kobor, M.S. Concordant and discordant DNA methylation signatures of aging in human blood and brain. Epigenetics Chromatin 2015, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Swindell, W.R. Genes and gene expression modules associated with caloric restriction and aging in the laboratory mouse. BMC Genom. 2009, 10, 585. [Google Scholar] [CrossRef] [Green Version]
- Langie, S.A.S.; Cameron, K.M.; Ficz, G.; Oxley, D.; Tomaszewski, B.; Gorniak, J.P.; Maas, L.M.; Godschalk, R.W.L.; Van Schooten, F.J.; Reik, W.; et al. The Ageing Brain: Effects on DNA Repair and DNA Methylation in Mice. Genes 2017, 8, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, S.M.; Fernandez-Lamo, I.; Schonig, K.; Fernandez Moya, S.M.; Ehses, J.; Schieweck, R.; Clementi, S.; Enkel, T.; Grothe, S.; von Bohlen Und Halbach, O.; et al. Forebrain-specific, conditional silencing of Staufen2 alters synaptic plasticity, learning, and memory in rats. Genome Biol. 2017, 18, 222. [Google Scholar] [CrossRef] [PubMed]
- Xu, X. DNA methylation and cognitive aging. Oncotarget 2015, 6, 13922–13932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blalock, E.M.; Chen, K.C.; Sharrow, K.; Herman, J.P.; Porter, N.M.; Foster, T.C.; Landfield, P.W. Gene microarrays in hippocampal aging: Statistical profiling identifies novel processes correlated with cognitive impairment. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 3807–3819. [Google Scholar] [CrossRef]
- Small, S.A.; Chawla, M.K.; Buonocore, M.; Rapp, P.R.; Barnes, C.A. Imaging correlates of brain function in monkeys and rats isolates a hippocampal subregion differentially vulnerable to aging. Proc. Natl. Acad. Sci. USA 2004, 101, 7181–7186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, S.N.; Barnes, C.A. Neural plasticity in the ageing brain. Nat. Rev. Neurosci. 2006, 7, 30–40. [Google Scholar] [CrossRef]
- Xu, X.; Zhan, M.; Duan, W.; Prabhu, V.; Brenneman, R.; Wood, W.; Firman, J.; Li, H.; Zhang, P.; Ibe, C.; et al. Gene expression atlas of the mouse central nervous system: Impact and interactions of age, energy intake and gender. Genome Biol. 2007, 8, R234. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Neff, F.; Dun, Z.; Hemmer, B.; Oertel, W.H.; Schlegel, J.; Hartmann, A. Gene expression profiles derived from single cells in human postmortem brain. Brain Res. Brain Res. Protoc. 2004, 13, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Jakovcevski, M.; Akbarian, S. Epigenetic mechanisms in neurological disease. Nat. Med. 2012, 18, 1194–1204. [Google Scholar] [CrossRef] [Green Version]
- Su, S.C.; Tsai, L.H. DNA methylation in cognition comes of age. Nat. Neurosci. 2012, 15, 1061–1062. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Xu, X. DNA Methyltransferases, DNA Methylation, and Age-Associated Cognitive Function. Int. J. Mol. Sci. 2018, 19, 1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, V.L.; Jones, P.A. DNA methylation decreases in aging but not in immortal cells. Science 1983, 220, 1055–1057. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Qian, H.; Ertl, R.; Astle, C.M.; Wang, G.G. Histone modifications change with age, dietary restriction and rapamycin treatment in mouse brain. Oncotarget 2015, 6, 15882–15890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Bang, E.; Jung, H.J.; Noh, S.G.; Yu, B.P.; Choi, Y.J.; Chung, H.Y. Anti-Aging Effects of Calorie Restriction (CR) and CR Mimetics Based on the Senoinflammation Concept. Nutrients 2020, 12, 422. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Wang, R.; Fok, W.C.; Coles, A.; Salmon, A.B.; Perez, V.I. Rapamycin and dietary restriction induce metabolically distinctive changes in mouse liver. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 410–420. [Google Scholar] [CrossRef] [Green Version]
- Fok, W.C.; Zhang, Y.; Salmon, A.B.; Bhattacharya, A.; Gunda, R.; Jones, D.; Ward, W.; Fisher, K.; Richardson, A.; Perez, V.I. Short-term treatment with rapamycin and dietary restriction have overlapping and distinctive effects in young mice. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 108–116. [Google Scholar] [CrossRef]
- Wilkinson, J.E.; Burmeister, L.; Brooks, S.V.; Chan, C.C.; Friedline, S.; Harrison, D.E.; Hejtmancik, J.F.; Nadon, N.; Strong, R.; Wood, L.K. Rapamycin slows aging in mice. Aging Cell 2012, 11, 675. [Google Scholar] [CrossRef] [Green Version]
- Unnikrishnan, A.; Kurup, K.; Salmon, A.B.; Richardson, A. Is Rapamycin a Dietary Restriction Mimetic? J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 4–13. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Rapamycin for longevity: Opinion article. Aging 2019, 11, 8048. [Google Scholar] [CrossRef]
- An, J.Y.; Kerns, K.A.; Ouellette, A.; Robinson, L.; Morris, H.D.; Kaczorowski, C.; Park, S.-I.; Mekvanich, T.; Kang, A.; McLean, J.S.; et al. Rapamycin rejuvenates oral health in aging mice. eLife 2020, 9, e54318. [Google Scholar] [CrossRef]
- Schinaman, J.M.; Rana, A.; Ja, W.W.; Clark, R.I.; Walker, D.W. Rapamycin modulates tissue aging and lifespan independently of the gut microbiota in Drosophila. Sci. Rep. 2019, 9, 7824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaeberlein, M.; Kennedy, B.K. Ageing: A midlife longevity drug? Nature 2009, 460, 331–332. [Google Scholar] [CrossRef] [PubMed]
- Garza-Lombo, C.; Gonsebatt, M.E. Mammalian Target of Rapamycin: Its Role in Early Neural Development and in Adult and Aged Brain Function. Front. Cell Neurosci. 2016, 10, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fok, W.C.; Bokov, A.; Gelfond, J.; Yu, Z.; Zhang, Y.; Doderer, M.; Chen, Y.; Javors, M.; Zhang, Y. Combined treatment of rapamycin and dietary restriction has a larger effect on the transcriptome and metabolome of liver. Aging Cell 2014, 13, 311–319. [Google Scholar] [CrossRef]
- Cole, J.J.; Robertson, N.A.; Rather, M.I.; Thomson, J.P.; McBryan, T.; Sproul, D.; Wang, T.; Brock, C.; Clark, W.; Ideker, T.; et al. Diverse interventions that extend mouse lifespan suppress shared age-associated epigenetic changes at critical gene regulatory regions. Genome Biol. 2017, 18, 58. [Google Scholar] [CrossRef] [Green Version]
- Tomazou, E.M.; Sheffield, N.C.; Schmidl, C.; Schuster, M.; Schonegger, A.; Datlinger, P.; Kubicek, S.; Bock, C.; Kovar, H. Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell Rep. 2015, 10, 1082–1095. [Google Scholar] [CrossRef] [Green Version]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Scholer, A.; van Nimwegen, E.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D.; et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490–495. [Google Scholar] [CrossRef]
- Qian, H.; Xu, X. Reduction in DNA methyltransferases and alteration of DNA methylation pattern associate with mouse skin ageing. Exp. Dermatol. 2014, 23, 357–359. [Google Scholar] [CrossRef]
- Guo, W.; Fiziev, P.; Yan, W.; Cokus, S.; Sun, X.; Zhang, M.Q.; Chen, P.Y.; Pellegrini, M. BS-Seeker2: A versatile aligning pipeline for bisulfite sequencing data. BMC Genom. 2013, 14, 774. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; Vanderplas, J. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2012, 12, 2825–2830. [Google Scholar]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Klopfenstein, D.V.; Zhang, L.; Pedersen, B.S.; Ramírez, F.; Warwick Vesztrocy, A.; Naldi, A.; Mungall, C.J.; Yunes, J.M.; Botvinnik, O.; Weigel, M.; et al. GOATOOLS: A Python library for Gene Ontology analyses. Sci. Rep. 2018, 8, 10872. [Google Scholar] [CrossRef]
- Lee, C. Gene Expression Profile of Aging and Its Retardation by Caloric Restriction. Science 1999, 285, 1390–1393. [Google Scholar] [CrossRef] [Green Version]
- Domcke, S.; Bardet, A.F.; Adrian Ginno, P.; Hartl, D.; Burger, L.; Schubeler, D. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature 2015, 528, 575–579. [Google Scholar] [CrossRef]
- Ianov, L.; Riva, A.; Kumar, A.; Foster, T.C. DNA Methylation of Synaptic Genes in the Prefrontal Cortex Is Associated with Aging and Age-Related Cognitive Impairment. Front. Aging Neurosci. 2017, 9, 249. [Google Scholar] [CrossRef]
- Ianov, L.; Rani, A.; Beas, B.S.; Kumar, A.; Foster, T.C. Transcription Profile of Aging and Cognition-Related Genes in the Medial Prefrontal Cortex. Front. Aging Neurosci. 2016, 8, 113. [Google Scholar] [CrossRef] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarkson-Townsend, D.A.; Everson, T.M.; Deyssenroth, M.A.; Burt, A.A.; Hermetz, K.E.; Hao, K.; Chen, J.; Marsit, C.J. Maternal circadian disruption is associated with variation in placental DNA methylation. PLoS ONE 2019, 14, e0215745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Haren, J.; Boudeau, J.; Schmidt, S.; Basu, S.; Liu, Z.; Lammers, D.; Demmers, J.; Benhari, J.; Grosveld, F.; Debant, A.; et al. Dynamic microtubules catalyze formation of navigator-TRIO complexes to regulate neurite extension. Curr Biol. 2014, 24, 1778–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hortsch, M. The L1 family of neural cell adhesion molecules: Old proteins performing new tricks. Neuron 1996, 17, 587–593. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, M.K.E.; Frotscher, M. Role of L1CAM for axon sprouting and branching. Cell Tissue Res. 2012, 349, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Gopisetty, G.; Ramachandran, K.; Singal, R. DNA methylation and apoptosis. Mol. Immunol. 2006, 43, 1729–1740. [Google Scholar] [CrossRef]
- Tang, H.; Shrager, J.; Goldman, D. Rapamycin protects aging muscle. Aging 2019, 11, 5868–5870. [Google Scholar] [CrossRef]
- Yoshino, Y.; Mori, T.; Yoshida, T.; Yamazaki, K.; Ozaki, Y.; Sao, T.; Funahashi, Y.; Iga, J.I.; Ueno, S.I. Elevated mRNA Expression and Low Methylation of SNCA in Japanese Alzheimer’s Disease Subjects. J. Alzheimers Dis. Jad 2016, 54, 1349. [Google Scholar] [CrossRef]
- Ozaki, Y.; Yoshino, Y.; Yamazaki, K.; Sao, T.; Mori, Y.; Ochi, S.; Yoshida, T.; Mori, T.; Iga, J.I.; Ueno, S.I. DNA methylation changes at TREM2 intron 1 and TREM2 mRNA expression in patients with Alzheimer’s disease. J. Psychiatr. Res. 2017, 92, 74–80. [Google Scholar] [CrossRef]
- Yamazaki, K.; Yoshino, Y.; Mori, T.; Yoshida, T.; Ozaki, Y.; Sao, T.; Mori, Y.; Ochi, S.; Iga, J.I.; Ueno, S.I. Gene Expression and Methylation Analysis of ABCA7 in Patients with Alzheimer’s Disease. J. Alzheimers Dis 2017, 57, 171–181. [Google Scholar] [CrossRef]
- Hadad, N.; Masser, D.R.; Logan, S.; Wronowski, B.; Mangold, C.A.; Clark, N.; Otalora, L.; Unnikrishnan, A.; Ford, M.M.; Giles, C.B.; et al. Absence of genomic hypomethylation or regulation of cytosine-modifying enzymes with aging in male and female mice. Epigenetics Chromatin 2016, 9, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unnikrishnan, A.; Hadad, N.; Masser, D.R.; Jackson, J.; Freeman, W.M.; Richardson, A. Revisiting the genomic hypomethylation hypothesis of aging. Ann. N. Y. Acad. Sci. 2018, 1418, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Slieker, R.C.; van Iterson, M.; Luijk, R.; Beekman, M.; Zhernakova, D.V.; Moed, M.H.; Mei, H.; van Galen, M.; Deelen, P.; Bonder, M.J.; et al. Age-related accrual of methylomic variability is linked to fundamental ageing mechanisms. Genome Biol. 2016, 17, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baribault, C.; Ehrlich, K.C.; Ponnaluri, V.K.C.; Pradhan, S.; Lacey, M.; Ehrlich, M. Developmentally linked human DNA hypermethylation is associated with down-modulation, repression, and upregulation of transcription. Epigenetics 2018, 13, 275–289. [Google Scholar] [CrossRef]
- Maston, G.A.; Evans, S.K.; Green, M.R. Transcriptional regulatory elements in the human genome. Annu Rev. Genom. Hum. Genet. 2006, 7, 29–59. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P.; Duan, W.; Lee, J.; Guo, Z. Suppression of brain aging and neurodegenerative disorders by dietary restriction and environmental enrichment: Molecular mechanisms. Mech. Ageing Dev. 2001, 122, 757–778. [Google Scholar] [CrossRef]
- Mattson, M.P. Methylation and acetylation in nervous system development and neurodegenerative disorders. Ageing Res. Rev. 2003, 2, 329–342. [Google Scholar] [CrossRef]
- Morrison, J.H.; Baxter, M.G. The ageing cortical synapse: Hallmarks and implications for cognitive decline. Nat. Rev. Neurosci. 2012, 13, 240–250. [Google Scholar] [CrossRef]
- Hadem, I.K.H.; Majaw, T.; Kharbuli, B.; Sharma, R. Beneficial effects of dietary restriction in aging brain. J. Chem. Neuroanat. 2019, 95, 123–133. [Google Scholar] [CrossRef]
- Reddy, P.H.; Beal, M.F. Amyloid β, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Oksenberg, N.; Ahituv, N. The role of AUTS2 in neurodevelopment and human evolution. Trends Genet. 2013, 29, 600–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, H.L.; Murphy, T.M.; Arseneault, L.; Caspi, A.; Moffitt, T.E.; Viana, J.; Hannon, E.; Pidsley, R.; Burrage, J.; Dempster, E.L.; et al. Methylomic analysis of monozygotic twins discordant for childhood psychotic symptoms. Epigenetics 2015, 10, 1014–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Togashi, H.; Sakisaka, T.; Takai, Y. Cell adhesion molecules in the central nervous system. Cell Adhes. Migr. 2009, 3, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rønn, L.C.; Berezin, V.; Bock, E. The neural cell adhesion molecule in synaptic plasticity and ageing. Int. J. Dev. Neurosci. 2000, 18, 193–199. [Google Scholar] [CrossRef]
- Zhang, Y.; Herman, B. Apoptosis and successful aging. Mech. Ageing Dev. 2002, 123, 563–565. [Google Scholar] [CrossRef]
- Friedrich, M.G.; Weisenberger DJCheng, J.C.; Chandrasoma, S.; Siegmund, K.D.; Gonzalgo, M.L.; Toma, M.I.; Huland, H.; Yoo, C.; Tsai, Y.C.; Nichols, P.W. Detection of methylated apoptosis-associated genes in urine sediments of bladder cancer patients. Clin. Cancer Res. 2004, 10, 7457–7465. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.H.; Liu, H.Q.; Cao, R.; Ji, A.L.; Zhang, L.; Wang, F.; Yang, R.H. Effects of dietary fish oil on learning function and apoptosis of hippocampal pyramidal neurons in streptozotocin-diabetic rats. Brain Res. 2012, 1457, 33–43. [Google Scholar] [CrossRef]
- Mercer, L.D.; Kelly, B.L.; Horne, M.K.; Beart, P.M. Dietary polyphenols protect dopamine neurons from oxidative insults and apoptosis: Investigations in primary rat mesencephalic cultures. Biochem. Pharm. 2005, 69, 339–345. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO | Name | #Gene | p-Value | Bonferroni |
|---|---|---|---|---|
| GO:0007218 | neuropeptide signaling pathway | 8 | 1.72 × 10−7 | 0.00204 |
| GO:0007268 | chemical synaptic transmission | 11 | 1.76 × 10−7 | 0.00208 |
| GO:0007399 | nervous system development | 19 | 2.52 × 10−7 | 0.00299 |
| GO:0016339 | calcium-dependent cell-cell adhesion via plasma membrane cell adhesion molecules | 6 | 3.34 × 10−7 | 0.00397 |
| GO:0007156 | homophilic cell adhesion via plasma membrane adhesion molecules | 10 | 4.69 × 10−7 | 0.00557 |
| GO:0006811 | ion transport | 18 | 5.57 × 10−7 | 0.00662 |
| GO:0001764 | neuron migration | 10 | 7.35 × 10−7 | 0.00873 |
| GO:0007411 | axon guidance | 14 | 8.90 × 10−7 | 0.0106 |
| GO:0044331 | cell-cell adhesion mediated by cadherin | 5 | 8.93 × 10−7 | 0.0106 |
| GO:0034332 | adherens junction organization | 5 | 2.42 × 10−6 | 0.0287 |
| GO | Name | #Gene | p-Value | Bonferroni |
|---|---|---|---|---|
| GO:0042590 | antigen processing and presentation of exogenous peptide antigen | 5 | 6.42 × 10−8 | 0.000762 |
| GO:0042270 | protection from natural killer cell mediated cytotoxicity | 5 | 6.42 × 10−8 | 0.000762 |
| GO:0042832 | defense response to protozoan | 11 | 7.56 × 10−8 | 0.000897 |
| GO:0007159 | leukocyte cell-cell adhesion | 10 | 9.21 × 10−8 | 0.00109 |
| GO:2000406 | positive regulation of T cell migration | 7 | 1.10 × 10−7 | 0.00131 |
| GO:0001516 | prostaglandin biosynthetic process | 8 | 1.10 × 10−7 | 0.00131 |
| GO:0016064 | immunoglobulin mediated immune response | 7 | 1.10 × 10−7 | 0.00131 |
| GO:0070098 | chemokine-mediated signaling pathway | 12 | 1.31 × 10−7 | 0.00155 |
| GO:0034341 | response to interferon-γ | 10 | 1.32 × 10−7 | 0.00157 |
| GO:0032088 | negative regulation of NF-kappaB transcription factor activity | 12 | 1.72 × 10−7 | 0.00204 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, Z.; Guo, X.; Qi, Y.; Li, P.; Liang, S.; Xu, X.; Shang, X. Dietary Restriction and Rapamycin Affect Brain Aging in Mice by Attenuating Age-Related DNA Methylation Changes. Genes 2022, 13, 699. https://doi.org/10.3390/genes13040699
Yin Z, Guo X, Qi Y, Li P, Liang S, Xu X, Shang X. Dietary Restriction and Rapamycin Affect Brain Aging in Mice by Attenuating Age-Related DNA Methylation Changes. Genes. 2022; 13(4):699. https://doi.org/10.3390/genes13040699
Chicago/Turabian StyleYin, Zhilei, Xinpeng Guo, Yang Qi, Pu Li, Shujun Liang, Xiangru Xu, and Xuequn Shang. 2022. "Dietary Restriction and Rapamycin Affect Brain Aging in Mice by Attenuating Age-Related DNA Methylation Changes" Genes 13, no. 4: 699. https://doi.org/10.3390/genes13040699
APA StyleYin, Z., Guo, X., Qi, Y., Li, P., Liang, S., Xu, X., & Shang, X. (2022). Dietary Restriction and Rapamycin Affect Brain Aging in Mice by Attenuating Age-Related DNA Methylation Changes. Genes, 13(4), 699. https://doi.org/10.3390/genes13040699