Abstract
5-methylcytosine (m5C) is a common post-transcriptional modification observed in a variety of RNAs. m5C has been demonstrated to be important in a variety of biological processes, including RNA structural stability and metabolism. Driven by the importance of m5C modification, many projects focused on the m5C sites prediction were reported before. To better understand the upstream and downstream regulation of m5C, we present a bioinformatics framework, m5CRegpred, to predict the substrate of m5C writer NSUN2 and m5C readers YBX1 and ALYREF for the first time. After features comparison, window lengths selection and algorism comparison on the mature mRNA model, our model achieved AUROC scores 0.869, 0.724 and 0.889 for NSUN2, YBX1 and ALYREF, respectively in an independent test. Our work suggests the substrate of m5C regulators can be distinguished and may help the research of m5C regulators in a special condition, such as substrates prediction of hyper- or hypo-expressed m5C regulators in human disease.
1. Introduction
Epitranscriptome is an emerging field in the past 10 years, and there are more than 170 types of RNA modifications identified [1]. 5-methylcytosine (m5C) is one of the prevalent RNA modifications, which has been found in most eukaryotes, prokaryotes, and archaea [2]. Biochemical research has revealed that m5C is abundant in tRNA and rRNA and serves a variety of molecular roles [3]. For example, m5C affects translation fidelity by altering the shape of rRNA to govern ribosome synthesis and processing [4]. The evolutionarily conserved m5C is responsible for maintaining the tertiary structures of tRNA [5]. Furthermore, new high-throughput investigations using bisulfite treatment or immunoprecipitation techniques have shown the presence of m5C on mRNA as well [6,7], which is associated with stability, export from nucleus [8], turnover [9], and translation [10] of mRNA.
Based on the developed LC–MS/MS technique, the estimated m5C/C ratio in human mRNA is about 0.02–0.09% [11]. With recent advances in genomics, at least 5 types of sequencing methods have been developed to reveal the epitranscriptome profile of m5C, including RNA-BisSeq [6], TAWO-seq [12], AZA-IP-seq [13], m5C-RIP-seq, and miCLIP-seq. These methods can be divided into two groups according to their principles: (a) chemical-dependent methods using bisulfite, peroxotungstate and 5-azacytidine in the first three methods, respectively; (b) the antibody-based methods using m5C-specific antibody and m5C-regulator antibody in the last two methods. Although the above methods provide capacity to detect m5C in transcriptome, comparing with the consensus DRACH [14] motif for m6A in most species, the exact m5C motif is still unknown. Using m5C-RIP-seq, multiple motifs for m5C were observed in Arabidopsis thaliana, including HACCR, CWUCUUC and CCDCCR [15], whereas m5C only showed an enrichment around CG-rich region in different species based on RNA-BisSeq [12,16,17,18].
Similar to methylation on DNA and protein, m5C is a reversible mark, which is deposited by methyltransferases and is removed by demethylases [10]. The members of NOP2/Sun RNA methyltransferase family are primary methyltransferases for m5C, including NSUN1, NSUN2, NSUN3, NSUN4, NSUN5, NSUN6, NSUN7. Some members from DNMT and TRDMT families are responsible for m5C deposition also. TET families and ALKBH1 regulated the demethylation of m5C on mRNA and tRNA which led to RNA degradation and mitochondrial activity, respectively [19]. Recent studies have reported that ALYREF and YBX1 are m5C binding proteins that can facilitate mRNA export [18] and stabilization [8] by recognizing m5C.
Although numerous effective bioinformatics studies for RNA modification sites prediction have been published in the epitranscriptomics field [20,21,22,23,24,25,26], none has focused on the substrate specificity of different m5C-related enzymes, such as methyltransferases (writers) and binding proteins (readers). In this work, we presented a bioinformatics framework “m5CRegpred” (which stands for m5C regulators substrate prediction, see Figure 1) based on machine learning and sequence-derived features to predict the substrate of m5C writers NSUN2 and readers YBX1 and ALFREF. The associations between diseases and m5C regulators have been reported before [27,28], especially that the hyper- or hypo-expression of NSUN2/YBX1/ALFREF were observed in multiple types of cancer [27,29,30,31,32]. This bioinformatics framework may help identify the substrate of each m5C regulators, which may provide another opportunity to understand their pathway in human diseases. The project code and training sequences are available at https://github.com/SXWuFJMU/m5CRegpred/ (accessed on 1 March 2022) and the supplement tables are available at https://github.com/SXWuFJMU/m5CRegpred/blob/main/Supplement%20Tables.zip (accessed on 1 March 2022).
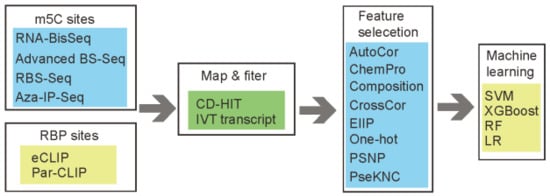
Figure 1.
The workflow for m5CRegpred. The methylation sites and RNA Binding proteins (RBP) sites were obtained from four and two types of sequencing techniques, respectively. Eight kinds of encoding methods were considered in the project.
2. Methods and Materials
2.1. The m5C Sites and Target Sites of the Enzymes
The transcriptome-wide m5C sites were extracted from the m6A-Atlas database [33], which were detected by four types of sequencing methods (Table 1). The sequences with 41 nt length and an m5C modification site at the center were generated to map with Par-CLIP [34] or eCLIP [35] data to identify the substrate of m5C regulators (Table 2). The substrates of m5C regulators were considered as the positive sites in the prediction. The unmethylated sites or unregulated sites from the same transcript with the positive sites were randomly selected as the negative sites, which keep the positive-to-negative ratio with 1:1. For each m5C regulator, the predictor was trained with 80% of the sites and the remaining 20% of sites were used for independent testing. To reduce the bias in the experiment, especially when selecting the polyA RNAs during library preparation, we built separate prediction models using full transcript data and mature mRNA data, respectively. In the mature mRNA predictor, only m5C sites located in exon regions are considered.

Table 1.
Base-resolution datasets of m5C sites.
Table 2.
Target sites of m5C regulators identified by Par-CLIP or eCLIP.
Considering the sequencing bias and non-specific binding of RNA modification antibody, the m5C sites identified from the IVT transcript [39] (in vitro transcribed RNA product consisted of only commercial NTPs, which should be free of modification) were used to filter false-positive sites and further improve the data quality. Additionally, the CD-HIT [40] software was used to remove redundant sequences with default parameters. As a result, sequence similarity is less than 85% in the dataset.
2.2. Sequence-Derived Features
Based on different physical and chemical properties, nucleotides can be decoded into different numeric vector or matrix. These encoding methods have been summarized in recent studies [41,42,43,44,45,46]. In this project, we selected eight popular methods in the RNA modification prediction field to identify the optimal features for substrate prediction: nucleic acid composition (CONPOSI), binary encoding method (ONE_HOT), position-specific nucleotide propensity (PSNP), electron-ion interaction pseudopotentials (EIIP), auto-correlation (autoCor), cross-correlation (crossCor), pseudo k-tupler composition (PseKNC) and chemical property (ChemProper).
2.3. Feature Description
Nucleic acid composition: Nucleic acid composition (CONPOSI) has been widely used in previous research [47]. In our study, dinucleotide frequencies were applied for sequence encoding, which can be presented as a 16-demensional feature vector (AA, AC, …, UU):
where the represents frequency of dinucleotide in the i-th sequence.
Binary encoding method: The nucleotide at each point in the flanking window is represented by four numeric values. The A, C, G, and U characters that fill the sequence termini were translated into binary vectors of (1,0,0,0), (0,1,0,0), (0,0,1,0), and (0,0,0,1), respectively.
Position-specific nucleotide propensity: The ‘position-specific nucleotide propensity based on single strand’ (PSTNPss) is a statistical method to encode the RNA sequences. In our study, the position-specific dinucleotide propensity was used, which contains 16 (i.e., 42) types of dinucleotides (e.g., ‘AA’, ‘AC’, ‘AG’, …, ‘UU’). Therefore, for an RNA sequence with L-bp length, the dinucleotide position specificity can be formulated as a matrix, where:
and represent the frequencies of the i-th dinucleotide (diN) at the j-th position appearing in positive dataset and negative dataset, respectively.
Electron-ion interaction pseudopotential: The EIIP method was proposed by Nair and Sreenadhan [48], which considers electron-ion interaction potential values between nucleotide. The EIIP values for each nucleic acid were shown blow:
In an RNA sequence, each nucleic acid will be replaced with its correspond EIIP value. For example, sequence ‘GCAU’ will be converted into a numeric vector (0.0806, 0.1340, 0.1260, 0.1335).
Auto-covariance and cross-covariance: the auto-covariance and cross-covariance were invented based on the physicochemical (PC) properties between two nucleotides [49]. In this work, we used ten types of PC to denote RNA, which can be formulated as a matrix:
where represents the i-th type of PC value of the j-th dinucleotide in the RNA sequence with L-bp length. Based on the PC matrix, the auto-covariance and cross-covariance can be calculated by following formulas, respectively:
where
The AC focuses on the correlation coefficient of the same physicochemical property between two subsequences, whereas CC considers the correlation coefficient between two subsequences with each belonging to a different PC property. The in this study equals to 39, which can capture more sequence information.
Pseudo k-tupler composition: PseKNC is the most widely used encoding method in the bioinformatic field, including protein, DNA and RNA prediction [50,51,52,53,54,55]. Several software/web servers/packages [41,42,43] have collected PseKNC methods in the suit. In this study, we directly used the PseKNC encoding method from ilearnplus web server to generate sequence-based features [43].
Chemical property: The sequence feature uses three unique structural chemical features to encode the nucleotide sequence: ring structures, functional groups, and hydrogen bonds. Adenine and cytosine have the amino group, whereas guanine and uracil have the keto group; adenine and guanine have two ring structures, whereas cytosine and uracil only have one; adenine and uracil can form two hydrogen bonds during hybridization, whereas guanine and cytosine can form three hydrogen bonds. Based on these chemical properties, the i-th nucleotide from sequence can be encoded by a vector
In other words, the A, C, G, U can be encoded as a vector (1,1,1), (0,1,0), (1,0,0) and (0,0,1), respectively.
2.4. Machine Learning Algorisms and Performance Evaluation
Machine learning algorithms have been widely used in many fields of biological research, such as miRNA target prediction, protein phosphorylation sites prediction, and achieved great performance in predicting RNA methylation sites. In this project, we used an R language interface of LIBSVM [56] to build Support Vector Machine (SVM) based predictors to compare encoding schemes and influence of sequence length. In addition, we compared multiple machine learning algorithms including SVM, Generalize Linear Model (GLM), Random Forest (RF), and XGBoost from R package caret [57] to identify a better algorithm for model construction. All default parameter in these functions were used to build predictors.
To validate the predictor performances, the five-fold cross-validation and independent test was employed for features selection purpose. The influences of sequence length and algorithms were evaluated by independent test only. The area under the receiver operating characteristic curve (AUROC) was calculated as the main performance evaluation metric. In addition to AUROC, the accuracy (ACC), sensitivity (Sn), and specificity (Sp) were calculated to measure the performance on algorithms comparison:
3. Results
3.1. Performances Based on Different Features
Recent studies have proven sequence-derived features are high reliability and effectiveness to reflect intrinsic relation to the targets. Here, we explored and compared eight different encoding methods for predicting the target specificity of m5C-regulators. After the CD-HIT filter, there are 269, 841, and 175 sequences considered as substrates of NSUN2, YBX1, ALYREF on mature mRNA model and 335, 1137, and 381 on full transcript model.
To identify the optimal features for the m5C-regulators prediction, the performance of 5-fold cross-validation on the training data (Table S1) and the independent test (Table 3) were both reported. In general, each feature achieved better performance on the full transcript model than the mature mRNA model, because the sequences of the exons are more conserved than sequences of introns which may have similar patterns. Among the eight encoding schemes, the PSNP methods achieved the best average performances on the regulator–substrate prediction, with AUROC scores of 0.869, 0.724, and 0.889 in independent tests of the NSUN2, YBX1, and ALYREF substrate prediction on the mature mRNA model. Although the “COMPOSITION” method had the best performances of YBX1 and ALYREF prediction on full transcript model, the performances are faint higher than PSNP method (0.764 of COMPOSITION vs. 0.763 of PSNP on YXB1 and 0.849 of COMPOSITION vs. 0.847 of PSNP on ALYREF). Additionally, the performances on mature mRNA model may reflect the actual prediction performances without overestimation due to polyA selection during library preparation [58]. Thus, the PSNP encoding method was selected to build predictors and further analysis.
Table 3.
Independent test with different features.
Additionally, except PSNP feature, two features with lower performance were combined randomly to test their performances (Table S2). Compared to PSNP feature, the EIIP–autoCovar combination features and EIIP–CONPOSI combination features achieved slight improvements (0.768 vs. 0.763; 0.849 vs. 0.847) for YBX1 and ALYREF substrate prediction, respectively, in full transcript model. These results suggest the PSNP may be the most appropriate feature for substrate prediction of m5C regulators.
3.2. Performances Based on Different Length Windows
The sequence windows length contains important sequence information and will affect the prediction performances [59,60], thus, we tried to optimize the length of the input sequences. The sequences with 21-, 31-, 41-, 51-, 61-, 71-, and 81-nt length and with an m5C modification site in the middle were tested to find the most promising prediction results (Figure 2 and Figure 3). On both the mature mRNA model and full transcript model, performances of NSUN2 and ALYREF substrate prediction were improving at the beginning, reaching the highest AUROC, and AUROC decreased as the length further increased. For YXB1 substrate prediction, the performances improved in a relatively steady manner and stabilized in the end as the length increased. Based on these results, the 51 nt and 51 nt for ALYREF, 71 nt and 61 nt for NSUN2, and 71 nt and 61 nt for YBX1 were selected on the mature mRNA model and full transcript model, respectively. These selected sequences can be freely accessed at https://github.com/SXWuFJMU/m5CRegpred/ (accessed on 1 March 2022).
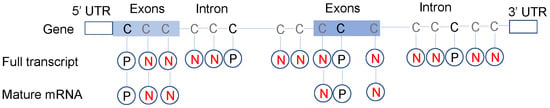
Figure 2.
Full transcript model and mature mRNA model. To select negative sites, the unmodified sites and methylated sites un-regulated by NSUN2/YBX1/ALYREF from the intron and exons were both considered in the full transcript model; whereas the mature mRNA model only considered sites from exons. As most captured sequences during library preparation are exons (mature mRNA) due to polyA selection, the performance of full transcript model will be overestimated.
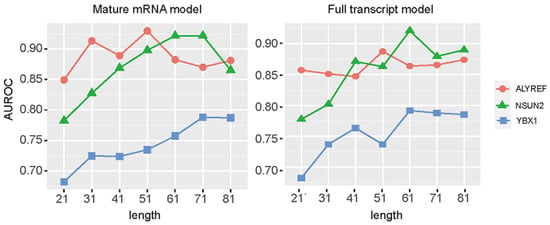
Figure 3.
Performance of different length windows with PSNP encoding method.
3.3. Performances Based on Different Machine Learning Algorithms
Although the SVM is the most popular algorithm on the RNA modification prediction filed [25,58,61,62,63,64,65,66,67], we also conducted a system comparison for the performances among SVM, RF, GLM, and XGBoost. The AUROC, accuracy, sensitivity, and specificity were calculated to measure the performance of predictors by independent test (Figure 4). In general, the performances were stable when the optimized sequence lengths were used among different machine learning algorithms, despite the SVM has the best performances.
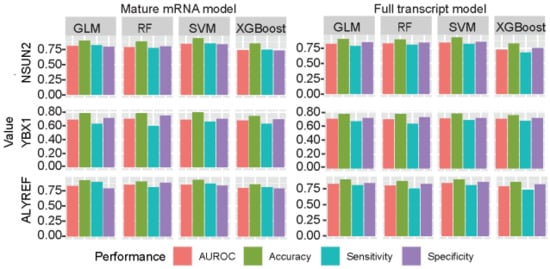
Figure 4.
Performance analysis on different machine learning algorithms. SVM represents for support vector machine, RF represents for random forest and GLMs represent for generalize linear model.
3.4. Data Interpretation
Although the exact motif for m5C modification sites is unknown, the motif of YBX1 substrate was identified as “CA(U/C)C” in human [68] before YBX1 is considered as an m5C reader. Further, Chen et al. [8] and Yang et al. [69] proved that YBX1 preferred to bind with “CA(U/C)m5C” rather than unmethylated “CA(U/C)C”, which suggests “CA(U/C)C” may be one motif for YBX1 dependent m5C. However, the motif of ALYREF is unclear. Another study based on NSUN2 knockout suggests “NGGG” is enriched among the NSUN2-dependent m5C sites [16]. In this project, after the YBX1 CLIP data mapping with m5C sites, there are 53 (3.36%, 53/1576) sequences contained “CA(U/C)C” motif. For NSUN2 data, there are 227 (55.6%, 227/408) sequences containing “NGGG”. The modification sites with potential motif were summarized in Supplementary Materials Table S3.
To better understand which sequences may contribute to the predictors, the motif among training data were analyzed by the STREME [70] from MEME suit. The most enriched motif for each regulator was presented in Figure 5. The results are similar to the previous studies, the GC-enriched sequences are around the m5C sites, regardless of whether there are substrates of NSUN2, YBX1, and ALYREF. Additionally, the motif for the YBX1 motif is insignificant, which may explain the lower performance of substrate prediction of YBX1 and suggest extra sequence-based features can be considered for the performance improvement. We also analyzed the motifs of the false-negative sites in the independent test. The false negative data of NSUN2 substrate are enriched in the GA-enriched sequences, whereas motifs for ALYREF false negative sites were different in full transcript and mature mRNA model.
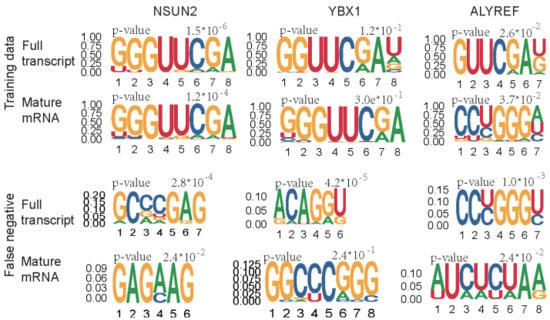
Figure 5.
Motif discovery for the training data andfalse-negative sites of independent test data. For the training data, only the positive sites were used for motif discovery. The motif with width of 4 bp to 8 bp was scanned by STREME. The different motifs of ALYREF in training data may be due to the different data size, which contained 296 sequences in the full transcript model whereas only 137 in mature mRNA model.
3.5. Case Study
The low resolution m5C profile on the breast epithelium cell line MCF10A generated by the m5C-meRIP technique was obtained from GSE53370 [71]. There are 1,744,029 cytosines located on the m5C peaks, and each cytosine was considered as the putative methylation sites. After prediction by m5CRegpred, 16,313 cytosines (Supplement Materials Table S4) were considered regulated by NSUN2 and recognized by at least one m5C reader with high confidence (probability > 95%). Among these results, cytosine located in gene PTPN2 (chromosome 18: 12789928), which is a tumor suppressor gene [72] with a low expression ratio in breast cancer [73], was a putative site regulated by NSUN2 and YBX1. The hypo-expression of NSUN2 were observed in the breast cancer [30], which may cause the low m5C level on mRNA. Although gene expression level of YBX1 is undifferentiated [74], less PTPN2 mRNA will be recognized by YBX1 due to the low methylation. Considering the YBX1 can stabilize mRNA [8], the impaired recognition by YBX1 will lead to the decay of PTPN2 and contribute to the development of breast cancer.
4. Discussion
In the past 10 years, RNA modifications-associated biological processes and molecular functions were widely explored to suggest the epi-transcriptome is an important layer in epigenetics regulation. The function and disease association of m5C were discussed also. Although the importance of m5C was proven, the attention on m5C modification is still less enthusiastic than m6A modification due to the lack of a dependable detecting method [7,16,60]. Here, we presented a bioinformatics work to show the substrates of m5C regulators can be distinguished by machine learning approaches, which provide a convenient and fast way on m5C relevant studies. In this study, we compared different encoding methods, length windows, and machine learning algorithms to build the optimal predictor (AUROC scores 0.869, 0.724, and 0.889 for NSUN2, YBX1, and ALYREF, respectively) on mature mRNA model. However, there are some limitations in the current study. The major defect is the bias of prediction results. The bias of result was considered in the site prediction field, such as using likelihood ratio (LR) to justify the probability. In these studies, the likelihood ratio was calculated by the probability of motif occurrence and the probability of observed RNA modification. Considering the motif of m5C is unclear (only can be summarized as the GC-enrich region) and the probability of observed RNA modification cannot be replaced with the probability of m5C regulated by NSUN2/YBX1/ALYREF, the bias is difficult to be calculated based on current knowledge.
Additionally, there are some shortcomings can be improved in further study. Firstly, although the sequence-derived features-based predictors have achieved acceptable performances, the advanced genomic features [75] should be considered to improve the performance in the future, especially for YBX1 substrate prediction. Secondly, the deep learning algorithms which were applied in the site prediction studies [76,77,78,79] recently, have better power than machine learning. Therefore, deep learning can be used to improve performance. Thirdly, the current prediction only focuses on one methyltransferase and two readers due to limited published dataset. More m5C regulators will be considered further once the sequencing results are released. Finally, some recent studies have suggested that RNA modification regulation is tissue-specific. Thus, the elaborate prediction with the tissues/cell lines specific should be considered in further research.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/genes13040677/s1, Supplementary Table S1: Cross-validation with different features, Table S2: Independent test with two features combination, Table S3: m5C sites with putative NSUN2/YBX1 motif, Table S4: MCF10A putative data.
Author Contributions
Conceptualization, H.S. and S.W.; methodology, Z.H. and J.X.; software, Z.H.; writing—original draft preparation, Z.H.; writing—review and editing, H.S.; supervision, H.S. and S.W.; project administration, S.W. All authors have read and agreed to the published version of the manuscript.
Funding
Scientific Research Foundation for Advanced Talents of Fujian Medical University (XRCZX2020012).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All relevant data is provided in the manuscript. Please contact at wushuxiang@fjmu.edu.cn for any raw data files and further analysis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Boccaletto, P.; Stefaniak, F.; Ray, A.; Cappannini, A.; Mukherjee, S.; Purta, E.; Kurkowska, M.; Shirvanizadeh, N.; Destefanis, E.; Groza, P.; et al. MODOMICS: A database of RNA modification pathways. 2021 update. Nucleic Acids Res. 2022, 50, D231–D235. [Google Scholar] [CrossRef] [PubMed]
- Trixl, L.; Lusser, A. The dynamic RNA modification 5-methylcytosine and its emerging role as an epitranscriptomic mark. Wiley Interdiscip. Rev. RNA 2019, 10, e1510. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Gao, C.C.; Gao, Y.; Yang, Y.; Shi, B.; Yu, J.L.; Lyu, C.; Sun, B.F.; Wang, H.L.; Xu, Y.; et al. OsNSUN2-Mediated 5-Methylcytosine mRNA Modification Enhances Rice Adaptation to High Temperature. Dev. Cell 2020, 53, 272–286.e277. [Google Scholar] [CrossRef] [PubMed]
- Heissenberger, C.; Liendl, L.; Nagelreiter, F.; Gonskikh, Y.; Yang, G.; Stelzer, E.M.; Krammer, T.L.; Micutkova, L.; Vogt, S.; Kreil, D.P.; et al. Loss of the ribosomal RNA methyltransferase NSUN5 impairs global protein synthesis and normal growth. Nucleic Acids Res. 2019, 47, 11807–11825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuorto, F.; Liebers, R.; Musch, T.; Schaefer, M.; Hofmann, S.; Kellner, S.; Frye, M.; Helm, M.; Stoecklin, G.; Lyko, F. RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat. Struct. Mol. Biol. 2012, 19, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.S.; Ma, H.L.; Yang, Y.; Lai, W.Y.; Sun, B.F.; Yang, Y.G. 5-Methylcytosine Analysis by RNA-BisSeq. Methods Mol. Biol. 2019, 1870, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Song, B.; Wei, Z.; Huang, D.; Zhang, Y.; Su, J.; de Magalhaes, J.P.; Rigden, D.J.; Meng, J.; Chen, K. m5C-Atlas: A comprehensive database for decoding and annotating the 5-methylcytosine (m5C) epitranscriptome. Nucleic Acids Res. 2022, 50, D196–D203. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, A.; Sun, B.F.; Yang, Y.; Han, Y.N.; Yuan, X.; Chen, R.X.; Wei, W.S.; Liu, Y.; Gao, C.C.; et al. 5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs. Nat. Cell Biol. 2019, 21, 978–990. [Google Scholar] [CrossRef]
- Cui, X.; Liang, Z.; Shen, L.; Zhang, Q.; Bao, S.; Geng, Y.; Zhang, B.; Leo, V.; Vardy, L.A.; Lu, T.; et al. 5-Methylcytosine RNA Methylation in Arabidopsis Thaliana. Mol. Plant 2017, 10, 1387–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.S.; Yang, W.L.; Zhao, Y.L.; Yang, Y.G. Dynamic transcriptomic m(5) C and its regulatory role in RNA processing. Wiley Interdiscip. Rev. RNA 2021, 12, e1639. [Google Scholar] [CrossRef] [PubMed]
- Abbasi-Moheb, L.; Mertel, S.; Gonsior, M.; Nouri-Vahid, L.; Kahrizi, K.; Cirak, S.; Wieczorek, D.; Motazacker, M.M.; Esmaeeli-Nieh, S.; Cremer, K.; et al. Mutations in NSUN2 cause autosomal-recessive intellectual disability. Am. J. Hum. Genet. 2012, 90, 847–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, F.; Bi, Y.; Siejka-Zielinska, P.; Zhou, Y.L.; Zhang, X.X.; Song, C.X. Bisulfite-free and base-resolution analysis of 5-methylcytidine and 5-hydroxymethylcytidine in RNA with peroxotungstate. Chem. Commun. 2019, 55, 2328–2331. [Google Scholar] [CrossRef] [Green Version]
- Khoddami, V.; Yerra, A.; Cairns, B.R. Experimental Approaches for Target Profiling of RNA Cytosine Methyltransferases. Methods Enzymol. 2015, 560, 273–296. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Chen, K.; Tang, Y.; Wei, Z.; Su, J.; de Magalhaes, J.P.; Rigden, D.J.; Meng, J. ConsRM: Collection and large-scale prediction of the evolutionarily conserved RNA methylation sites, with implications for the functional epitranscriptome. Brief Bioinform 2021, 22, bbab088. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Perrera, V.; Saplaoura, E.; Apelt, F.; Bahin, M.; Kramdi, A.; Olas, J.; Mueller-Roeber, B.; Sokolowska, E.; Zhang, W.; et al. m(5)C Methylation Guides Systemic Transport of Messenger RNA over Graft Junctions in Plants. Curr. Biol. CB 2019, 29, 2465–2476.e2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.; Chen, W.; Liu, J.; Gu, N.; Zhang, R. Genome-wide identification of mRNA 5-methylcytosine in mammals. Nat. Struct. Mol. Biol. 2019, 26, 380–388. [Google Scholar] [CrossRef]
- David, R.; Burgess, A.; Parker, B.; Li, J.; Pulsford, K.; Sibbritt, T.; Preiss, T.; Searle, I.R. Transcriptome-Wide Mapping of RNA 5-Methylcytosine in Arabidopsis mRNAs and Noncoding RNAs. Plant Cell 2017, 29, 445–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Yang, Y.; Sun, B.F.; Chen, Y.S.; Xu, J.W.; Lai, W.Y.; Li, A.; Wang, X.; Bhattarai, D.P.; Xiao, W.; et al. 5-methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Res. 2017, 27, 606–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Liu, F.; Chen, W.; Miao, H.; Liang, H.; Liao, Z.; Zhang, Z.; Zhang, B. The role of RNA m(5)C modification in cancer metastasis. Int. J. Biol. Sci. 2021, 17, 3369–3380. [Google Scholar] [CrossRef]
- Liu, L.; Song, B.; Chen, K.; Zhang, Y.; de Magalhaes, J.P.; Rigden, D.J.; Lei, X.; Wei, Z. WHISTLE server: A high-accuracy genomic coordinate-based machine learning platform for RNA modification prediction. Methods 2021. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Chen, K.; Meng, J. WHISTLE: A Functionally Annotated High-Accuracy Map of Human m(6)A Epitranscriptome. Methods Mol. Biol. 2021, 2284, 519–529. [Google Scholar] [CrossRef]
- Chen, X.; Xiong, Y.; Liu, Y.; Chen, Y.; Bi, S.; Zhu, X. m5CPred-SVM: A novel method for predicting m5C sites of RNA. BMC Bioinform. 2020, 21, 489. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Li, X.; Ding, H.; Xu, L.; Xiang, H. Prediction of m5C Modifications in RNA Sequences by Combining Multiple Sequence Features. Mol. Ther. Nucleic Acids 2020, 21, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Chai, D.; Jia, C.; Zheng, J.; Zou, Q.; Li, F. Staem5: A novel computational approachfor accurate prediction of m5C site. Mol. Ther. Nucleic Acids 2021, 26, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Song, B.; Ma, J.; Song, Y.; Zhang, S.Y.; Tang, Y.; Wu, X.; Wei, Z.; Chen, K.; Su, J.; et al. Bioinformatics approaches for deciphering the epitranscriptome: Recent progress and emerging topics. Comput. Struct. Biotechnol. J. 2020, 18, 1587–1604. [Google Scholar] [CrossRef]
- Song, B.; Tang, Y.; Chen, K.; Wei, Z.; Rong, R.; Lu, Z.; Su, J.; de Magalhaes, J.P.; Rigden, D.J.; Meng, J. m7GHub: Deciphering the location, regulation and pathogenesis of internal mRNA N7-methylguanosine (m7G) sites in human. Bioinformatics 2020, 36, 3528–3536. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Xie, X.; Han, G.; Zhang, T.; Li, Y.; Li, Y.; Yin, R.; Wang, Q.; Zhang, T.; Wang, P.; et al. YBX1 is required for maintaining myeloid leukemia cell survival by regulating BCL2 stability in an m6A-dependent manner. Blood 2021, 138, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, M.; Zhao, Y.L.; Yang, Y.; Yang, Y.G. RNA methylations in human cancers. Semin. Cancer Biol. 2021, 75, 97–115. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, C.; Tong, X.; Chen, S.; Hu, X.; Pan, B.; Sun, X.; Chen, Z.; Shi, X.; Hu, Y.; et al. NSUN2 modified by SUMO-2/3 promotes gastric cancer progression and regulates mRNA m5C methylation. Cell Death Dis. 2021, 12, 842. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Pan, J.; Wang, H.; Du, X.; Xu, Y.; Wang, Z.; Chen, D. Prognostic Significance and Tumor Immune Microenvironment Heterogenicity of m5C RNA Methylation Regulators in Triple-Negative Breast Cancer. Front. Cell Dev. Biol. 2021, 9, 657547. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Shen, C.; Miao, R.; Wang, J.Z.; Cao, M.D.; Zhang, Y.S.; Shi, L.H.; Zhao, G.H.; Wang, M.H.; Wu, L.S.; et al. RNA methyltransferase NSUN2 promotes gastric cancer cell proliferation by repressing p57(Kip2) by an m(5)C-dependent manner. Cell Death Dis. 2020, 11, 270. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.Z.; Zhu, W.; Han, J.; Yang, X.; Zhou, R.; Lu, H.C.; Yu, H.; Yuan, W.B.; Li, P.C.; Tao, J.; et al. The role of the HIF-1alpha/ALYREF/PKM2 axis in glycolysis and tumorigenesis of bladder cancer. Cancer Commun. 2021, 41, 560–575. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Chen, K.; Song, B.; Ma, J.; Wu, X.; Xu, Q.; Wei, Z.; Su, J.; Liu, G.; Rong, R.; et al. m6A-Atlas: A comprehensive knowledgebase for unraveling the N6-methyladenosine (m6A) epitranscriptome. Nucleic Acids Res. 2021, 49, D134–D143. [Google Scholar] [CrossRef] [PubMed]
- Danan, C.; Manickavel, S.; Hafner, M. PAR-CLIP: A Method for Transcriptome-Wide Identification of RNA Binding Protein Interaction Sites. Methods Mol. Biol. 2022, 2404, 167–188. [Google Scholar] [CrossRef]
- Van Nostrand, E.L.; Pratt, G.A.; Shishkin, A.A.; Gelboin-Burkhart, C.; Fang, M.Y.; Sundararaman, B.; Blue, S.M.; Nguyen, T.B.; Surka, C.; Elkins, K.; et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 2016, 13, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Khoddami, V.; Yerra, A.; Mosbruger, T.L.; Fleming, A.M.; Burrows, C.J.; Cairns, B.R. Transcriptome-wide profiling of multiple RNA modifications simultaneously at single-base resolution. Proc. Natl. Acad. Sci. USA 2019, 116, 6784–6789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoddami, V.; Cairns, B.R. Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nat. Biotechnol. 2013, 31, 458–464. [Google Scholar] [CrossRef] [Green Version]
- Frankish, A.; Diekhans, M.; Jungreis, I.; Lagarde, J.; Loveland, J.E.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Armstrong, J.; Barnes, I.; et al. Gencode 2021. Nucleic Acids Res. 2021, 49, D916–D923. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, T.; Chen, H.X.; Xie, Y.Y.; Chen, L.Q.; Zhao, Y.L.; Liu, B.D.; Jin, L.; Zhang, W.; Liu, C.; et al. Systematic calibration of epitranscriptomic maps using a synthetic modification-free RNA library. Nat. Methods 2021, 18, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, F.; Wang, X.; Chen, J.; Fang, L.; Chou, K.C. Pse-in-One: A web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015, 43, W65–W71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Gao, X.; Zhang, H. BioSeq-Analysis2.0: An updated platform for analyzing DNA, RNA and protein sequences at sequence level and residue level based on machine learning approaches. Nucleic Acids Res. 2019, 47, e127. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhao, P.; Li, C.; Li, F.; Xiang, D.; Chen, Y.Z.; Akutsu, T.; Daly, R.J.; Webb, G.I.; Zhao, Q.; et al. iLearnPlus: A comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic Acids Res. 2021, 49, e60. [Google Scholar] [CrossRef]
- Liu, B. BioSeq-Analysis: A platform for DNA, RNA and protein sequence analysis based on machine learning approaches. Brief Bioinform 2019, 20, 1280–1294. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhao, P.; Li, F.; Marquez-Lago, T.T.; Leier, A.; Revote, J.; Zhu, Y.; Powell, D.R.; Akutsu, T.; Webb, G.I.; et al. iLearn: An integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data. Brief Bioinform 2020, 21, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, X.; Brooker, J.; Lin, H.; Zhang, L.; Chou, K.C. PseKNC-General: A cross-platform package for generating various modes of pseudo nucleotide compositions. Bioinformatics 2015, 31, 119–120. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, Y.; Ning, Q.; Zhang, H.; Ji, J.; Yin, M. Identifying N(6)-methyladenosine sites using extreme gradient boosting system optimized by particle swarm optimizer. J. Theor. Biol. 2019, 467, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.S.; Sreenadhan, S.P. A coding measure scheme employing electron-ion interaction pseudopotential (EIIP). Bioinformation 2006, 1, 197–202. [Google Scholar] [PubMed]
- Zou, H.; Yang, F.; Yin, Z. Identifying N7-methylguanosine sites by integrating multiple features. Biopolymers 2022, 113, e23480. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Weng, F.; Huang, D.S.; Chou, K.C. iRO-3wPseKNC: Identify DNA replication origins by three-window-based PseKNC. Bioinformatics 2018, 34, 3086–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.H.; Deng, E.Z.; Xu, L.Q.; Ding, H.; Lin, H.; Chen, W.; Chou, K.C. iNuc-PseKNC: A sequence-based predictor for predicting nucleosome positioning in genomes with pseudo k-tuple nucleotide composition. Bioinformatics 2014, 30, 1522–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, C.Q.; Zhang, Z.Y.; Zhu, X.J.; Lin, Y.; Chen, W.; Tang, H.; Lin, H. iTerm-PseKNC: A sequence-based tool for predicting bacterial transcriptional terminators. Bioinformatics 2019, 35, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Yang, H.; Ding, H.; Lin, H.; Chen, W.; Chou, K.C. iDNA6mA-PseKNC: Identifying DNA N(6)-methyladenosine sites by incorporating nucleotide physicochemical properties into PseKNC. Genomics 2019, 111, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Yang, F.; Huang, D.S.; Chou, K.C. iPromoter-2L: A two-layer predictor for identifying promoters and their types by multi-window-based PseKNC. Bioinformatics 2018, 34, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.D.; Huang, Y.; Zhang, Z.Y.; Zhao, Y.W.; Wang, D.; Chen, W.; Chou, K.C.; Lin, H. iLoc-lncRNA: Predict the subcellular location of lncRNAs by incorporating octamer composition into general PseKNC. Bioinformatics 2018, 34, 4196–4204. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-C.; Lin, C.-J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. (TIST) 2011, 2, 1–27. [Google Scholar] [CrossRef]
- Kuhn, M. Building Predictive Models inRUsing thecaretPackage. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Wei, Z.; Zhang, Q.; Wu, X.; Rong, R.; Lu, Z.; Su, J.; de Magalhaes, J.P.; Rigden, D.J.; Meng, J. WHISTLE: A high-accuracy map of the human N6-methyladenosine (m6A) epitranscriptome predicted using a machine learning approach. Nucleic Acids Res. 2019, 47, e41. [Google Scholar] [CrossRef] [Green Version]
- Zou, Q.; Xing, P.; Wei, L.; Liu, B. Gene2vec: Gene subsequence embedding for prediction of mammalian N (6)-methyladenosine sites from mRNA. RNA 2019, 25, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Z.; Huang, D.; Song, B.; Chen, K.; Song, Y.; Liu, G.; Su, J.; Magalhaes, J.P.; Rigden, D.J.; Meng, J. Attention-based multi-label neural networks for integrated prediction and interpretation of twelve widely occurring RNA modifications. Nat. Commun. 2021, 12, 4011. [Google Scholar] [CrossRef]
- Chen, W.; Feng, P.; Song, X.; Lv, H.; Lin, H. iRNA-m7G: Identifying N(7)-methylguanosine Sites by Fusing Multiple Features. Mol. Ther. Nucleic Acids 2019, 18, 269–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Song, X.; Lv, H.; Lin, H. iRNA-m2G: Identifying N(2)-methylguanosine Sites Based on Sequence-Derived Information. Mol. Ther. Nucleic Acids 2019, 18, 253–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Tang, H.; Ye, J.; Lin, H.; Chou, K.C. iRNA-PseU: Identifying RNA pseudouridine sites. Mol. Ther. Nucleic Acids 2016, 5, e332. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Ding, H.; Yang, H.; Chen, W.; Lin, H.; Chou, K.C. iRNA-PseColl: Identifying the Occurrence Sites of Different RNA Modifications by Incorporating Collective Effects of Nucleotides into PseKNC. Mol. Ther. Nucleic Acids 2017, 7, 155–163. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Feng, P.; Ding, H.; Lin, H.; Chou, K.C. iRNA-Methyl: Identifying N(6)-methyladenosine sites using pseudo nucleotide composition. Anal. Biochem. 2015, 490, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.; Yang, H.; Ding, H.; Lin, H.; Chou, K.C. iRNA-3typeA: Identifying Three Types of Modification at RNA's Adenosine Sites. Mol. Ther. Nucleic Acids 2018, 11, 468–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Song, B.; Tang, Y.; Wei, Z.; Xu, Q.; Su, J.; de Magalhaes, J.P.; Rigden, D.J.; Meng, J. RMDisease: A database of genetic variants that affect RNA modifications, with implications for epitranscriptome pathogenesis. Nucleic Acids Res. 2021, 49, D1396–D1404. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.J.; Mu, S.R.; Heiner, M.; Fu, X.; Cao, L.J.; Gong, X.F.; Bindereif, A.; Hui, J. YB-1 binds to CAUC motifs and stimulates exon inclusion by enhancing the recruitment of U2AF to weak polypyrimidine tracts. Nucleic Acids Res. 2012, 40, 8622–8636. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, L.; Han, X.; Yang, W.L.; Zhang, M.; Ma, H.L.; Sun, B.F.; Li, A.; Xia, J.; Chen, J.; et al. RNA 5-Methylcytosine Facilitates the Maternal-to-Zygotic Transition by Preventing Maternal mRNA Decay. Mol. Cell 2019, 75, 1188–1202.e1111. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L. STREME: Accurate and versatile sequence motif discovery. Bioinformatics 2021, 37, 2834–2840. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Panneerdoss, S.; Timilsina, S.; Zhu, J.; Mohammad, T.A.; Lu, Z.L.; de Magalhaes, J.P.; Chen, Y.; Rong, R.; Huang, Y.; et al. Topological Characterization of Human and Mouse m(5)C Epitranscriptome Revealed by Bisulfite Sequencing. Int. J. Genom. 2018, 2018, 1351964. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Kim, P.; Mitra, R.; Zhao, J.; Zhao, Z. TSGene 2.0: An updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res. 2016, 44, D1023–D1031. [Google Scholar] [CrossRef] [PubMed]
- Veenstra, C.; Karlsson, E.; Mirwani, S.M.; Nordenskjold, B.; Fornander, T.; Perez-Tenorio, G.; Stal, O. The effects of PTPN2 loss on cell signalling and clinical outcome in relation to breast cancer subtype. J. Cancer Res. Clin. Oncol. 2019, 145, 1845–1856. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; He, S.; Guo, F.; Zou, Q. HSM6AP: A high-precision predictor for the Homo sapiens N6-methyladenosine (m^6 A) based on multiple weights and feature stitching. RNA Biol. 2021, 18, 1882–1892. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Zhang, L.; Chen, H.; Huang, D.; Song, B. m6A-Maize: Weakly supervised prediction of m(6)A-carrying transcripts and m(6)A-affecting mutations in maize (Zea mays). Methods 2021. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Song, B.; Wei, J.; Su, J.; Coenen, F.; Meng, J. Weakly supervised learning of RNA modifications from low-resolution epitranscriptome data. Bioinformatics 2021, 37, i222–i230. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, P.; Li, F.; Wang, Y.; Smith, A.I.; Webb, G.I.; Akutsu, T.; Baggag, A.; Bensmail, H.; Song, J. Comprehensive review and assessment of computational methods for predicting RNA post-transcriptional modification sites from RNA sequences. Brief Bioinform 2020, 21, 1676–1696. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Guo, X.; Jin, P.; Chen, J.; Xiang, D.; Song, J.; Coin, L.J.M. Porpoise: A new approach for accurate prediction of RNA pseudouridine sites. Brief Bioinform 2021, 22, bbab245. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).