

Full-Length Transcriptome Analysis of the Halophyte Nitraria sibirica Pall

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Salt Treatment
2.2. Nucleic Acid Extraction and SMRT Sequencing
2.3. Preprocessing of Raw Data
2.4. CDS, SSR, Transcription Factors (TFs), LncRNA Analysis
2.5. Functional Annotation
2.6. Alternative Splice Detected
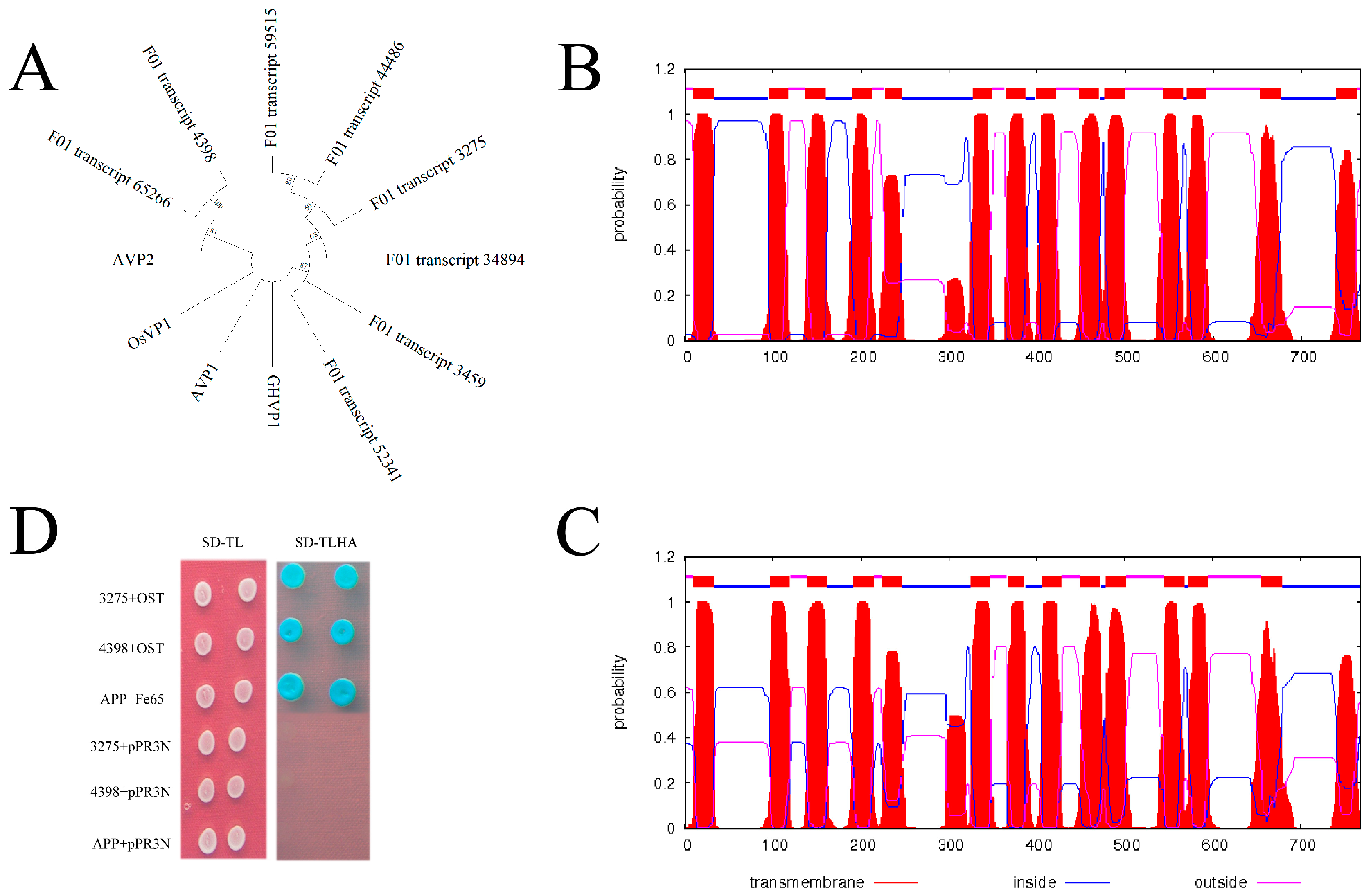
2.7. Phylogenetic and Subcellular Localization Analysis
2.8. Function Analysis of H+-PPase
3. Results
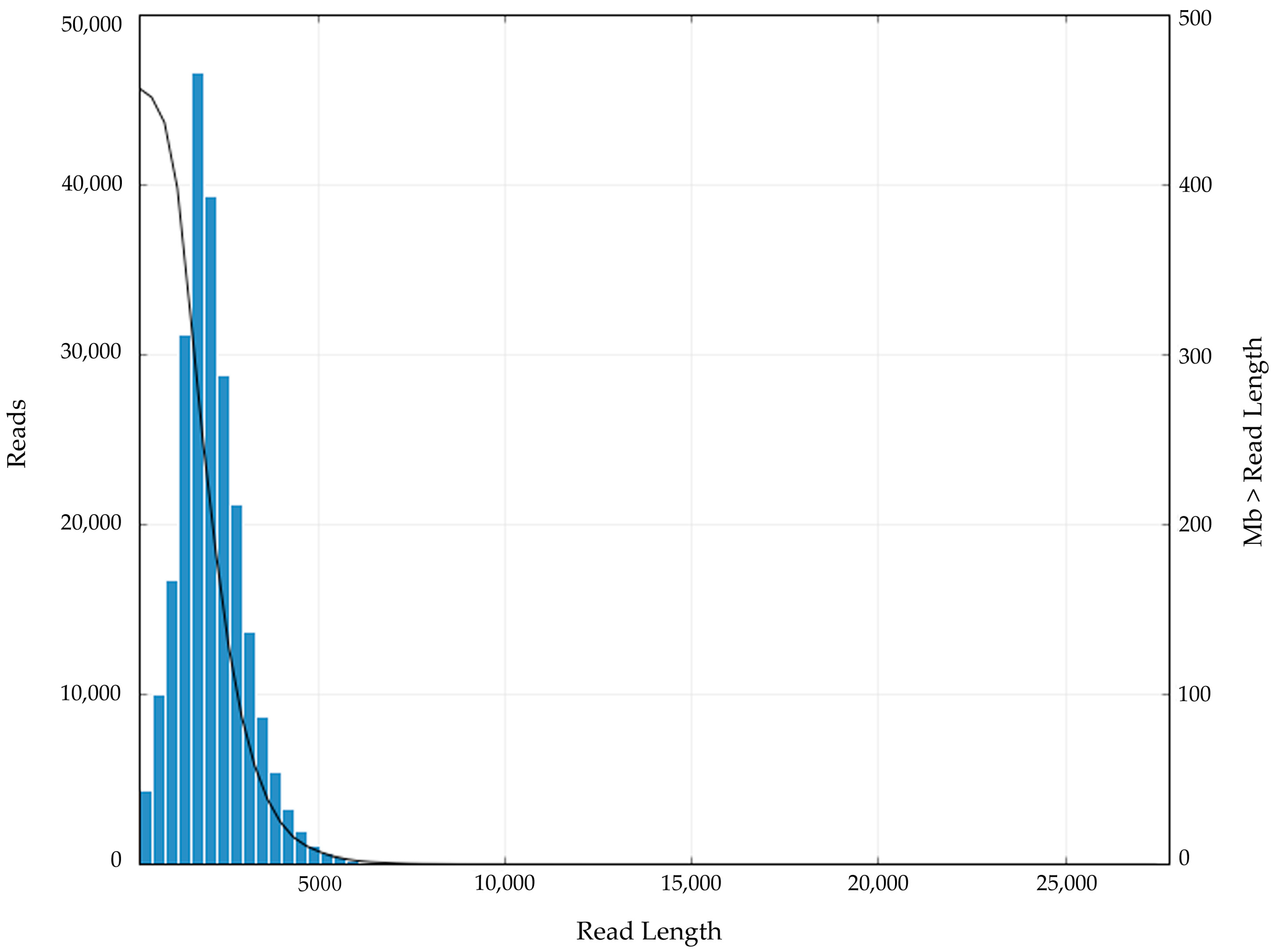
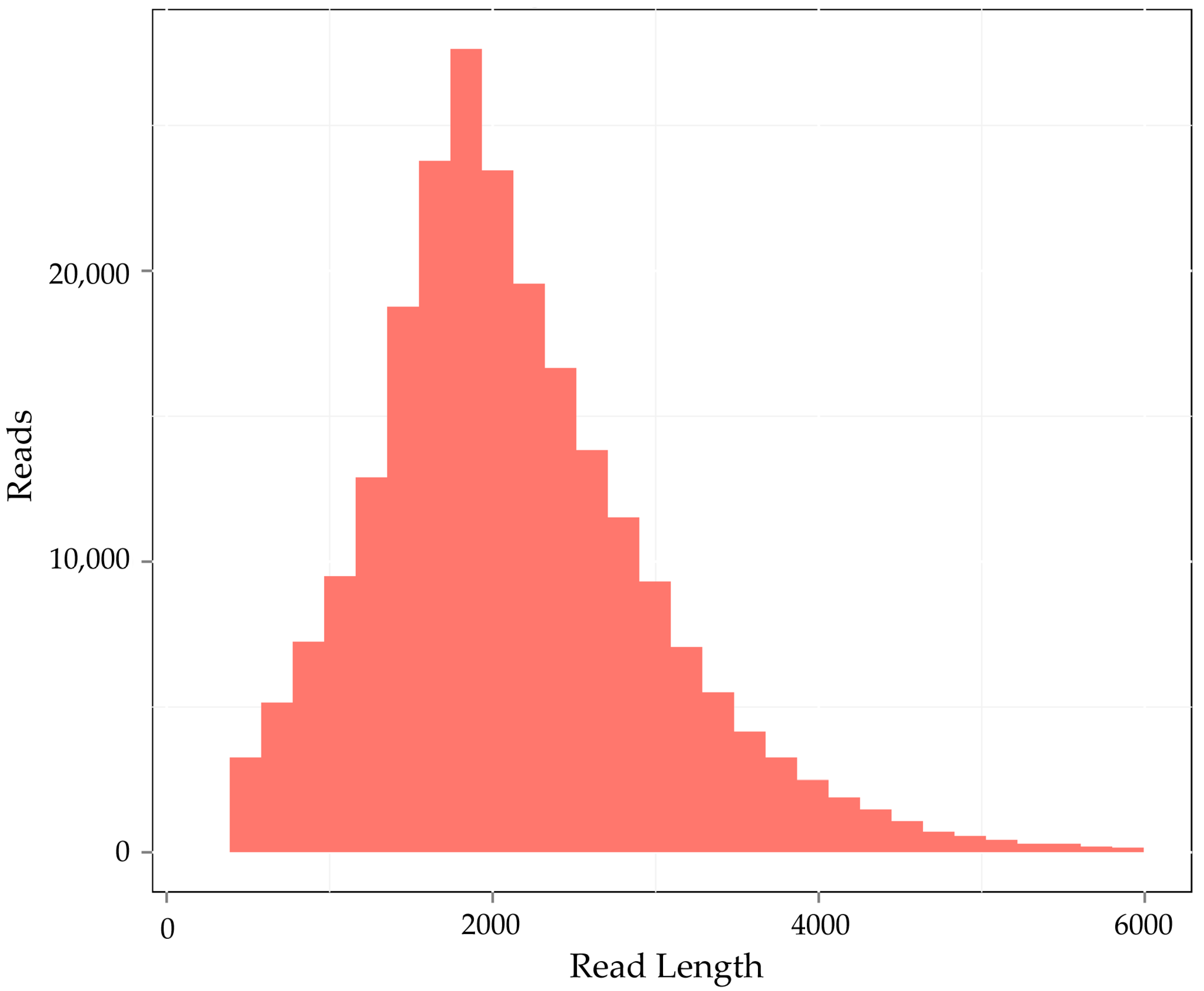
3.1. The Sequencing Data Analysis of N. sibirica
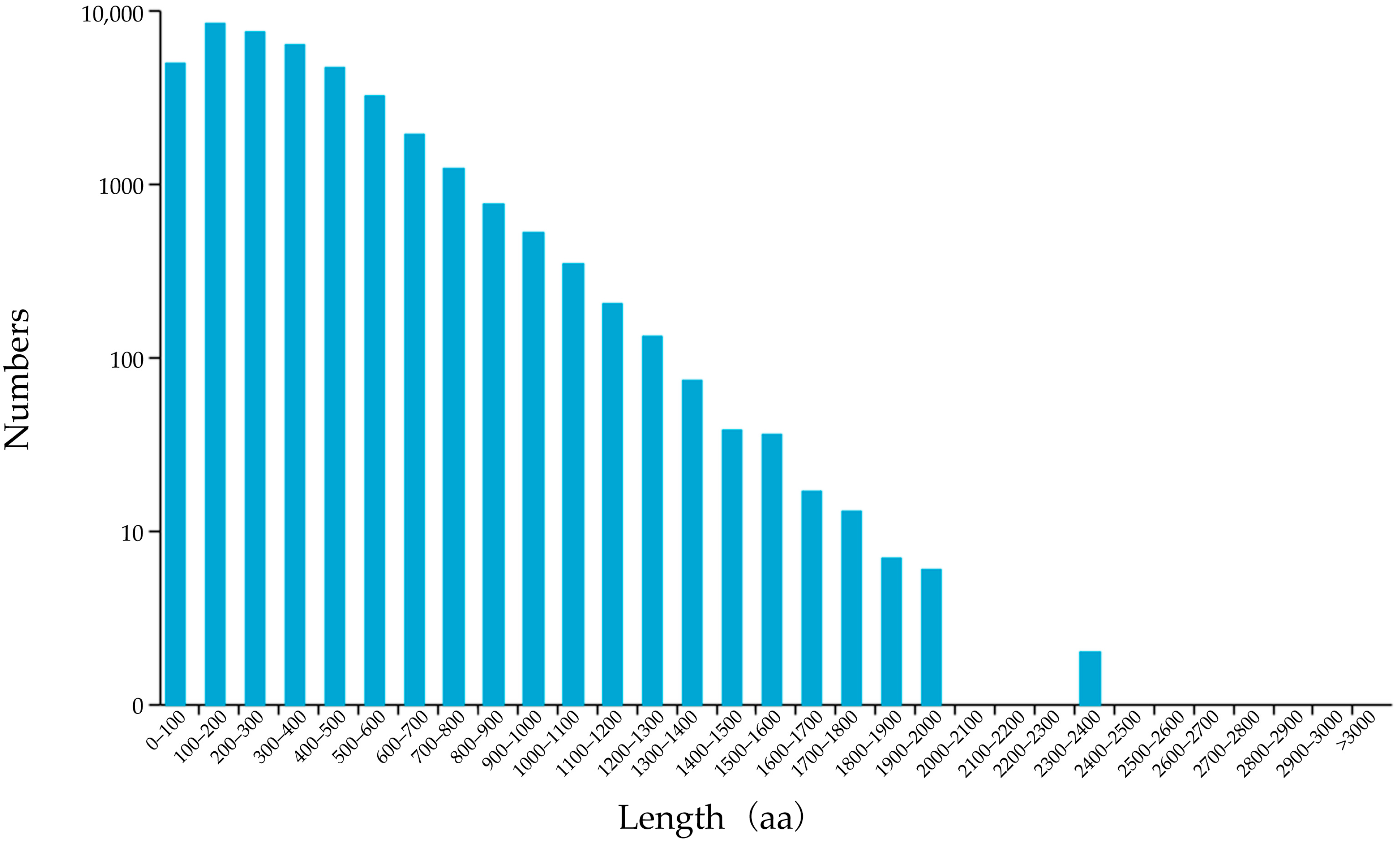
3.2. Prediction of Coding Sequences
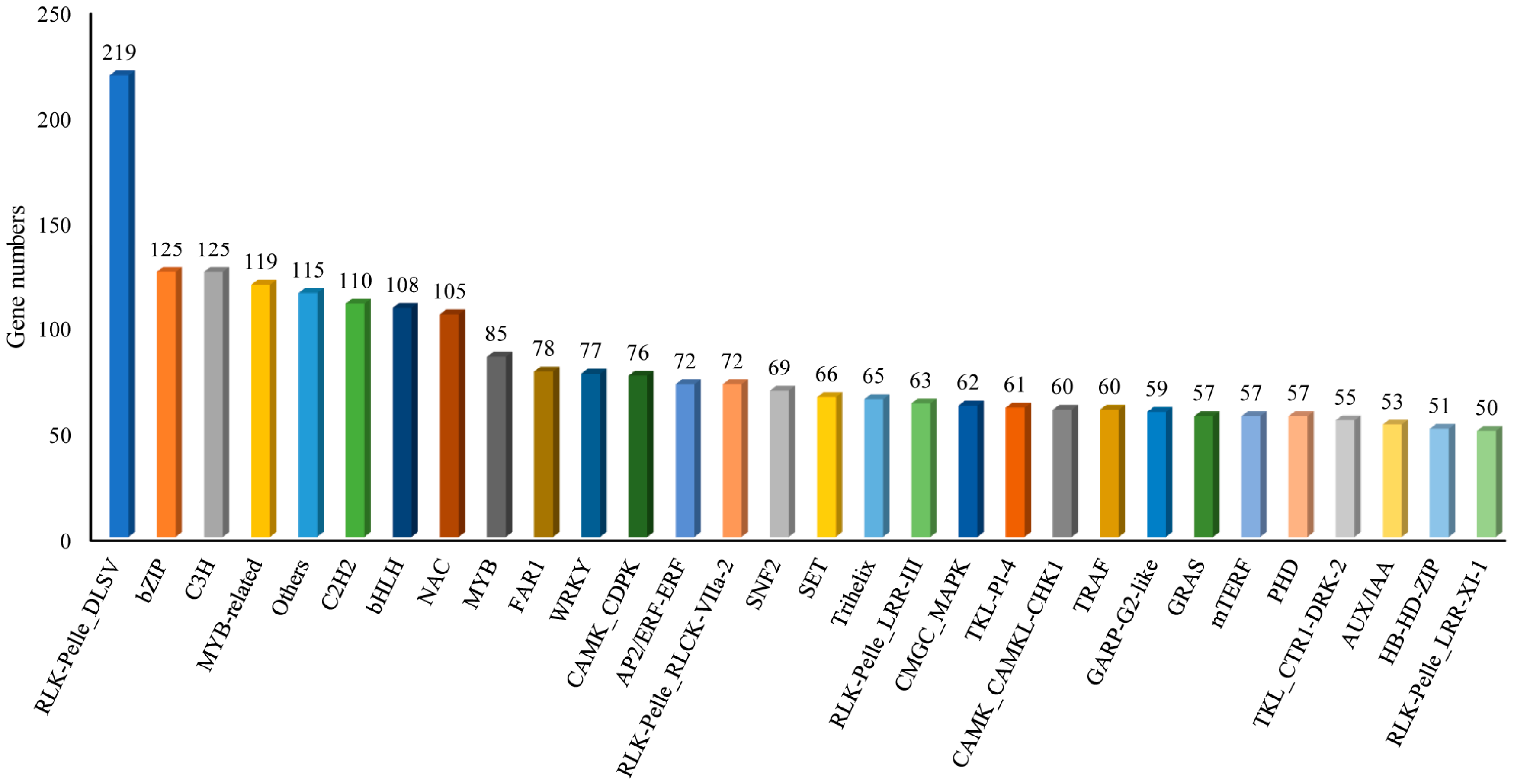
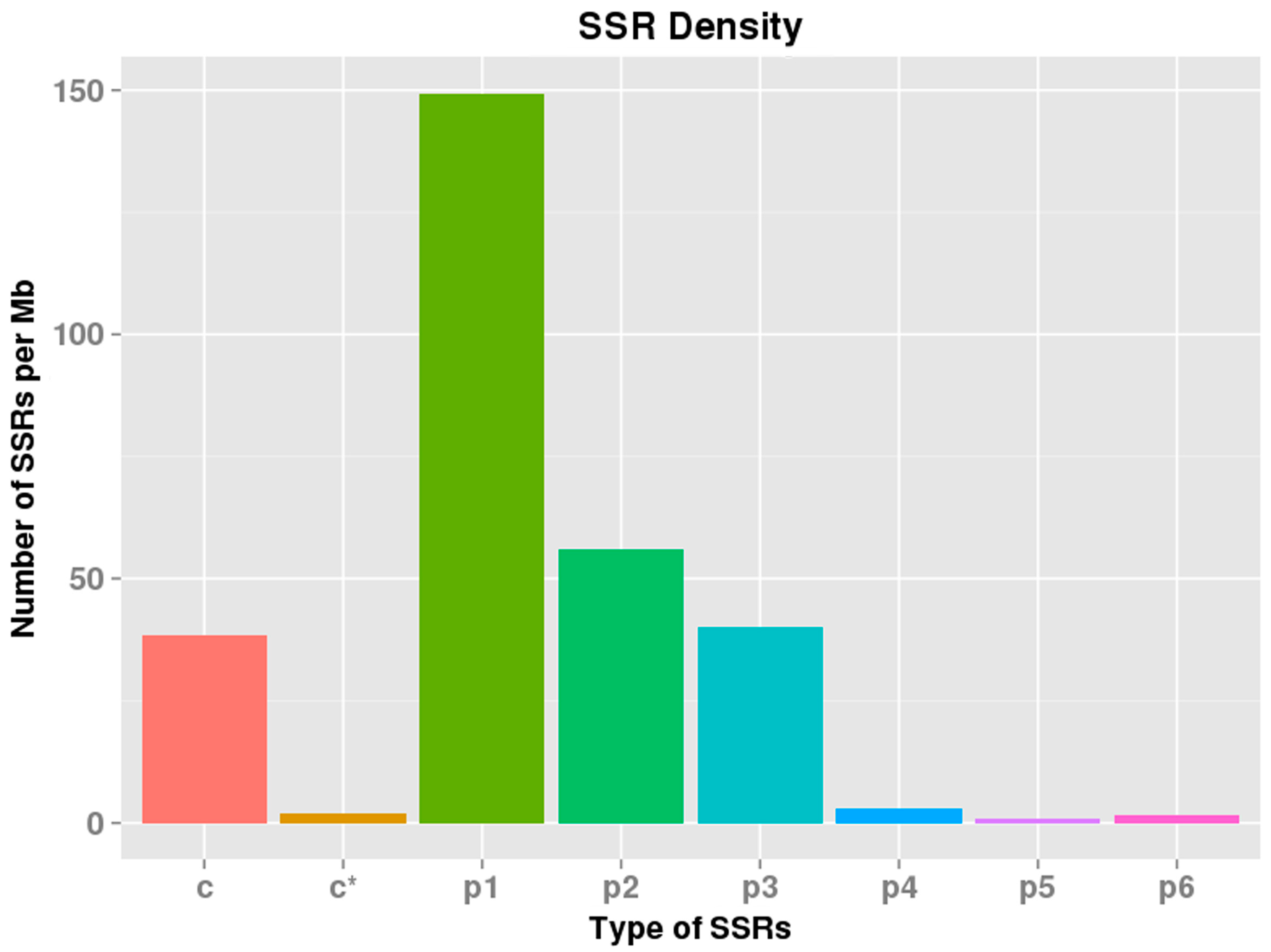
3.3. SSR Discovery
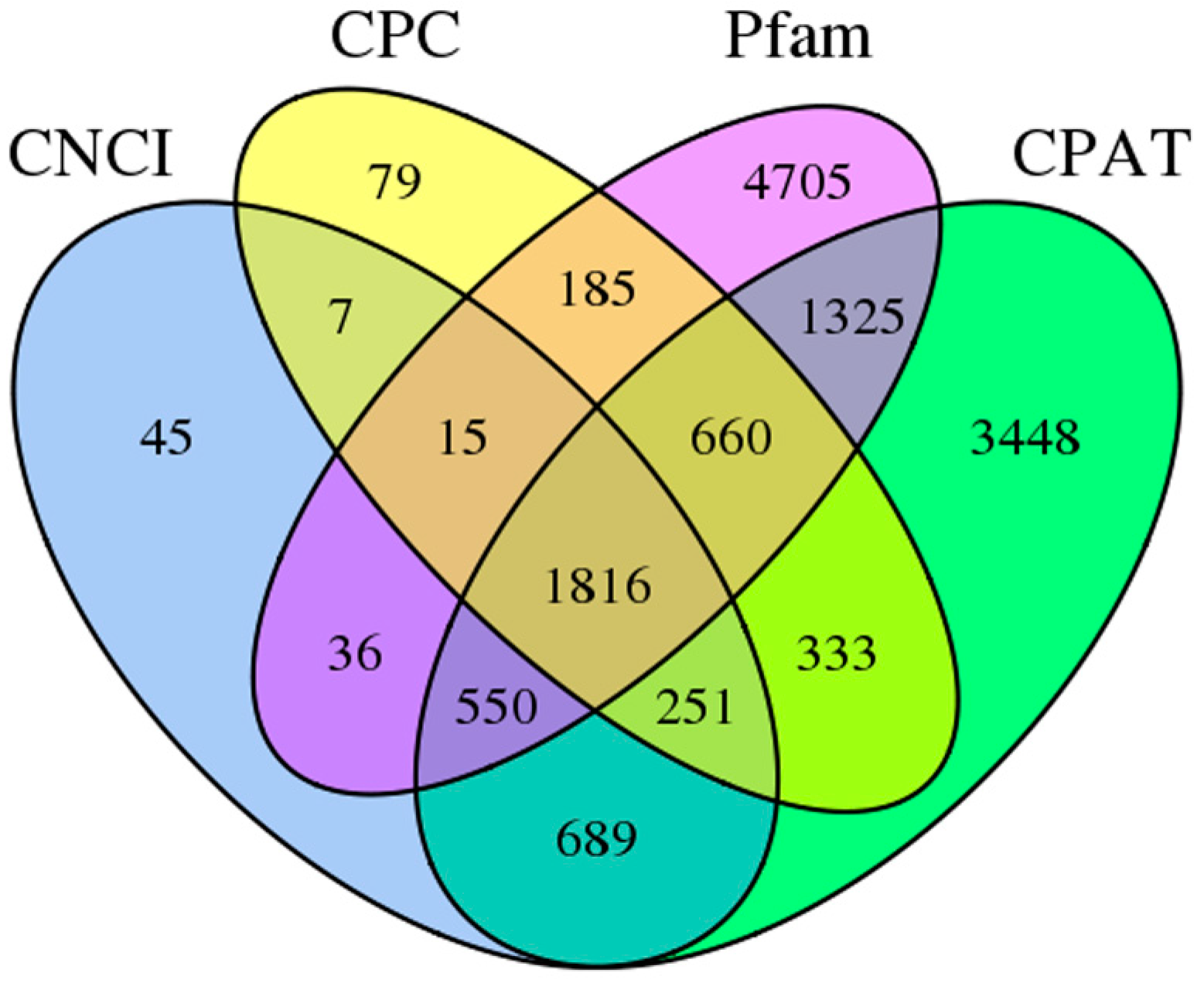
3.4. LncRNA Prediction and Alternative Splicing Analysis
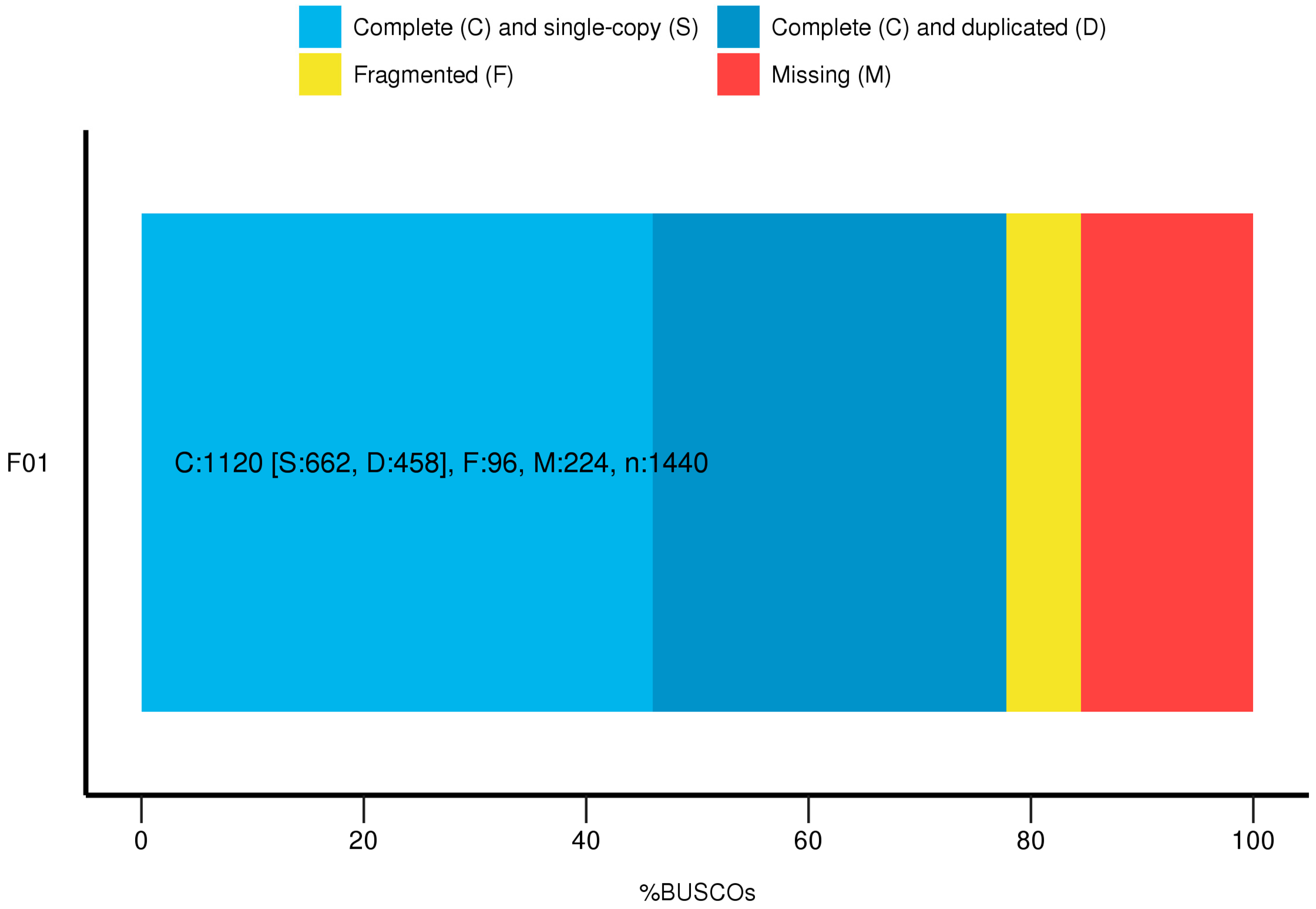
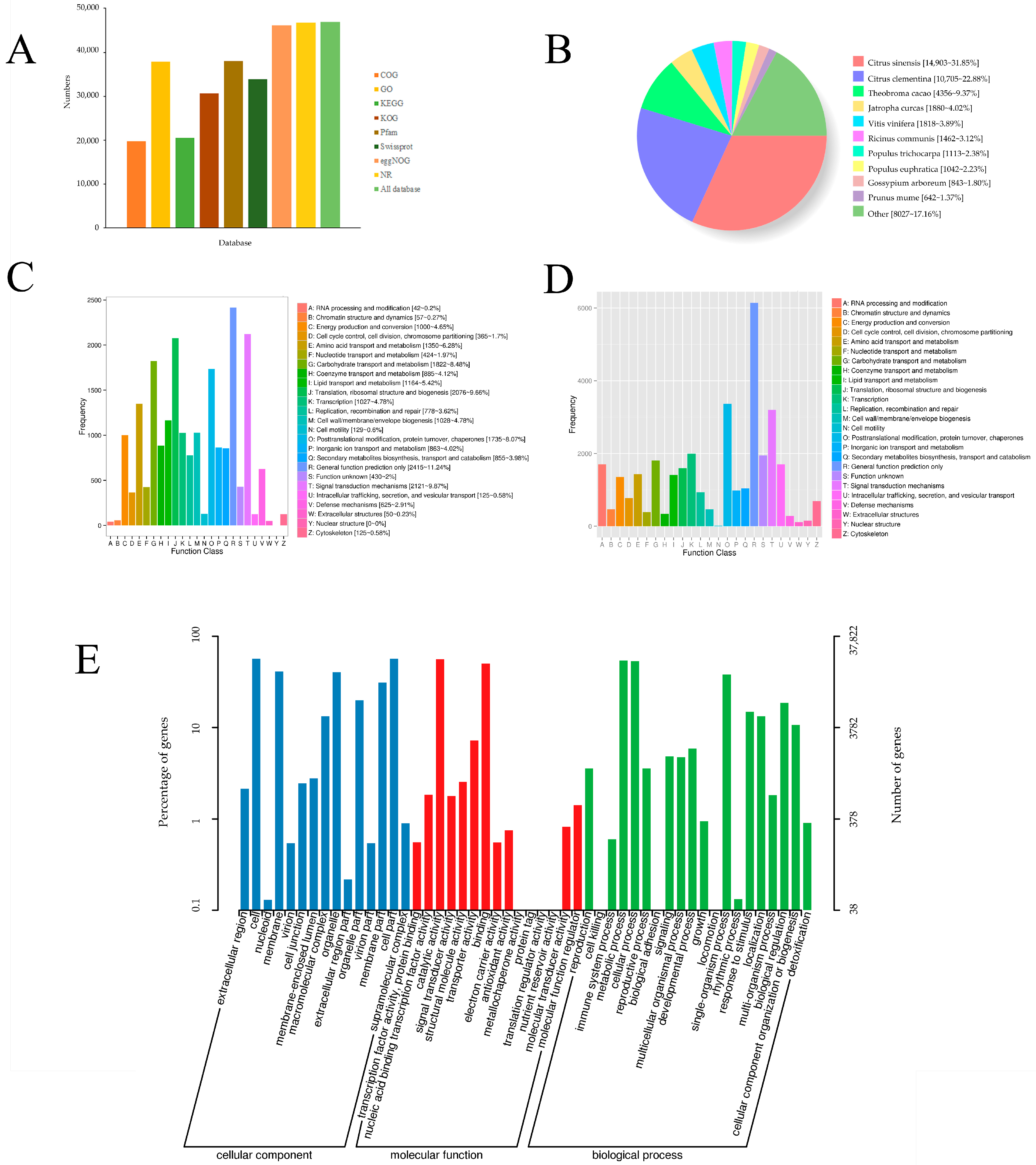
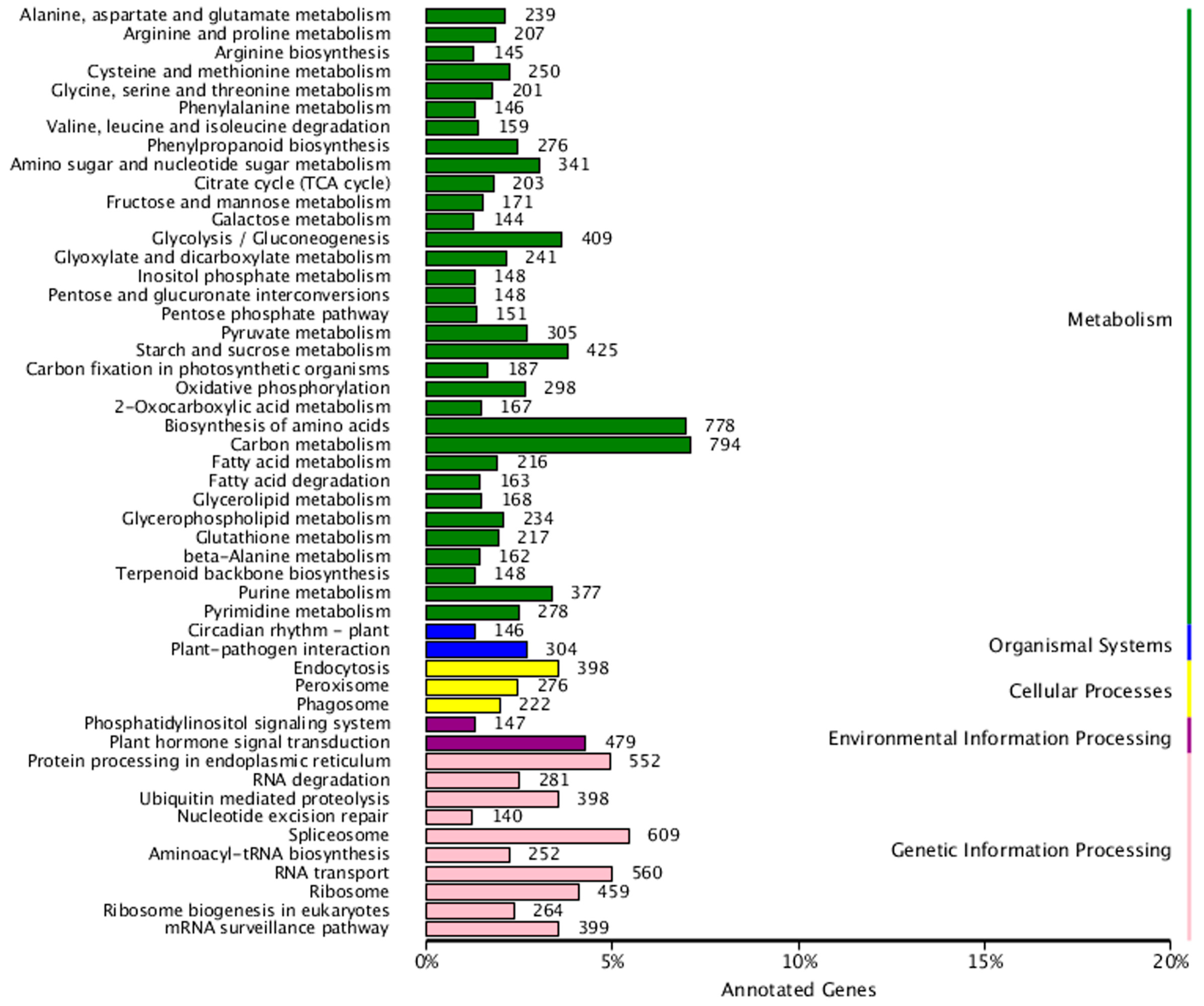
3.5. Functional Annotation
3.6. Characterization of H+-PPase Genes Based on Full-Length Transcriptome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tang, X.; Zhang, H.; Sergey, S.; Li, H.; Yang, X.; Zhang, H. Tissue Tolerance Mechanisms Conferring Salinity Tolerance in a Halophytic Perennial Species Nitraria sibirica Pall. Tree Physiol. 2020, 7, 1264–1277. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.B.; Zhao, H.D.; Fu, Z.L.; Chen, D.F.; Lu, Y. Antioxidant and Anticomplement Compounds Isolated from Nitraria sibirica Fruit by High-Speed Counter-Current Chromatography. Pharmacogn. Mag. 2018, 14, 541. [Google Scholar]
- Su, W.; Zhang, W. Study on Extraction of Red Pigment from Nitraria sibirica Fruit and Its Stability. J. Tianjin Agric. Coll. 2002, 4, 5–11. [Google Scholar]
- Tang, X.; Yang, X.; Li, H.; Zhang, H. Maintenance of K+/Na+ Balance in the Roots of Nitraria sibirica Pall. in Response to NaCl Stress. Forests 2018, 9, 601. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Li, H.; Yang, X.; Wu, H.; Liu, X.; Zhang, H. Effect of NaCl Stress on Root Growth and K+/Na+ Balance of Nitraria sibirica Pall. Seedlings. J. Northwest A F Univ. Nat. Sci. Ed. 2019, 8, 83–89. [Google Scholar]
- Chen, G.; Wang, C.; Chen, J. Osmosis-Regulating Substance and Cation Contents in Nitraria sibirica Pall. Test-Tube Plantlets with NaCl Stress. Acta Bot. Boreali-Occident. Sin. 2009, 6, 1233–1239. [Google Scholar]
- Rina, S.A.; Chen, G. Effect of Exogenous Spermidine on Antioxidant Enzyme System in Leaves of Nitraria sibirica Pall. Seedlings under Salt Stress. Acta Bot. Boreali-Occident. Sin. 2013, 2, 352–356. [Google Scholar]
- Bashir, K.; Matsui, A.; Rasheed, S.; Seki, M. Recent Advances in the Characterization of Plant Transcriptomes in Response to Drought, Salinity, Heat, and Cold Stress. F1000Research 2019, 8, 658. [Google Scholar] [CrossRef] [Green Version]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, X.; Chory, J. The Arabidopsis Transcriptome Responds Specifically and Dynamically to High Light Stress. Cell Rep. 2019, 29, 4186–4199. [Google Scholar] [CrossRef] [Green Version]
- Nobori, T.; Velásquez, A.C.; Wu, J.; Kvitko, B.H.; Kremer, J.M.; Wang, Y.; He, S.Y.; Tsuda, K. Transcriptome Landscape of a Bacterial Pathogen under Plant Immunity. Proc. Natl. Acad. Sci. USA 2018, 115, 3055–3064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petre, B.; Lorrain, C.; Stukenbrock, E.H.; Duplessis, S. Host-Specialized Transcriptome of Plant-Associated Organisms. Curr. Opin. Plant Biol. 2020, 56, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Tang, X.; Yang, X.; Zhang, H. Comprehensive Transcriptome and Metabolome Profiling Reveal Metabolic Mechanisms of Nitraria sibirica Pall. to Salt Stress. Sci. Rep. 2021, 11, 12878. [Google Scholar] [CrossRef] [PubMed]
- Ardui, S.; Ameur, A.; Vermeesch, J.R.; Hestand, M.S. Single Molecule Real-Time (SMRT) Sequencing Comes of Age: Applications and Utilities for Medical Diagnostics. Nucleic Acids Res. 2018, 46, 2159–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-Read Human Genome Sequencing and Its Applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef]
- Nakano, K.; Shiroma, A.; Shimoji, M.; Tamotsu, H.; Ashimine, N.; Ohki, S.; Shinzato, M.; Minami, M.; Nakanishi, T.; Teruya, K.; et al. Advantages of Genome Sequencing by Long-Read Sequencer Using SMRT Technology in Medical Area. Hum. Cell 2017, 30, 149–161. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Zaman, S.; Hu, S.; Che, S. Single-Molecule Long-Read Sequencing of Purslane (Portulaca oleracea) and Differential Gene Expression Related with Biosynthesis of Unsaturated Fatty Acids. Plants 2021, 10, 655. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, H.; Kohnen, M.V.; Prasad, K.V.S.K.; Gu, L.; Reddy, A.S.N. Analysis of Transcriptome and Epitranscriptome in Plants Using PacBio Iso-Seq and Nanopore-Based Direct RNA Sequencing. Front. Genet. 2019, 10, 253. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Chen, L.; Chen, C.; An, X.; Zhang, Y.; Wang, Y.; Li, Q. Full-Length Transcriptome Analysis of Phytolacca americana and Its Congener P. icosandra and Gene Expression Normalization in Three Phytolaccaceae Species. BMC Plant Biol. 2020, 20, 396. [Google Scholar] [CrossRef]
- Sharon, D.; Tilgner, H.; Grubert, F.; Snyder, M. A Single-Molecule Long-Read Survey of the Human Transcriptome. Nat. Biotechnol. 2013, 31, 1009–1014. [Google Scholar] [CrossRef]
- Chao, Y.; Yuan, J.; Li, S.; Jia, S.; Han, L.; Xu, L. Analysis of Transcripts and Splice Isoforms in Red Clover (Trifolium pratense L.) by Single-Molecule Long-Read Sequencing. BMC Plant Biol. 2018, 18, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Xiang, Y.; Xiong, Y.; Lin, Z.; Xue, Y.; Mao, M.; Sun, L.; Zhou, Y.; Li, X.; Huang, Z. SMRT Sequencing Analysis Reveals the Full-Length Transcripts and Alternative Splicing Patterns in Ananas comosus Var. bracteatus. PeerJ 2019, 7, e7062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Wang, J.; Wang, Z.; Li, X.; Kan, J.; Lin, J. SMRT Sequencing of Full-Length Transcriptome of Birch-Leaf Pear (Pyrus betulifolia Bunge) under Drought Stress. J. Genet. 2021, 100, 29. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Xu, D.; Zhuo, Z.; Hu, J.; Lu, B. SMRT Sequencing of the Full-Length Transcriptome of the Rhynchophorus ferrugineus (Coleoptera: Curculionidae). PeerJ 2020, 8, 9133. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Tang, X.; Zhu, J.; Yang, X.; Zhang, H. De Novo Transcriptome Characterization, Gene Expression Profiling and Ionic Responses of Nitraria sibirica Pall. under Salt Stress. Forests 2017, 8, 211. [Google Scholar] [CrossRef] [Green Version]
- Yuan, F.; Guo, J.; Shabala, S.; Wang, B. Reproductive Physiology of Halophytes: Current Standing. Front. Plant Sci. 2018, 9, 1954. [Google Scholar] [CrossRef]
- Rahman, M.M.; Mostofa, M.G.; Keya, S.S.; Siddiqui, M.N.; Ansary, M.M.U.; Das, A.K.; Rahman, M.A.; Tran, L.S.-P. Adaptive Mechanisms of Halophytes and Their Potential in Improving Salinity Tolerance in Plants. Int. J. Mol. Sci. 2021, 22, 10733. [Google Scholar] [CrossRef]
- Madritsch, S.; Burg, A.; Sehr, E.M. Comparing de Novo Transcriptome Assembly Tools in Di- and Autotetraploid Non-Model Plant Species. BMC Bioinform. 2021, 22, 146. [Google Scholar] [CrossRef]
- Zhu, L.; Lu, L.; Yang, L.; Hao, Z.; Chen, J.; Cheng, T. The Full-Length Transcriptome Sequencing and Identification of Na+/H+ Antiporter Genes in Halophyte Nitraria tangutorum Bobrov. Genes 2021, 12, 836. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Li, A.; Yu, B.; Li, S. Interplay between MiRNAs and LncRNAs: Mode of Action and Biological Roles in Plant Development and Stress Adaptation. Comput. Struct. Biotechnol. J. 2021, 19, 2567–2574. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, Y.; Chen, X.; Chen, Y. Plant Noncoding RNAs: Hidden Players in Development and Stress Responses. Annu. Rev. Cell Dev. Biol. 2019, 35, 407–431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, W.; Zhu, W.; Dong, J.; Cheng, Y.; Yin, Z.; Shen, F. Mechanisms and Functions of Long Non-Coding RNAs at Multiple Regulatory Levels. Int. J. Mol. Sci. 2019, 20, 5573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Li, Y.; Chu, L.; Kuang, X.; Song, J.; Sun, C. Full-Length Sequencing of Ginkgo Transcriptomes for an in-Depth understanding of Flavonoid and Terpenoid Trilactone Biosynthesis. Gene 2020, 758, 144961. [Google Scholar] [CrossRef] [PubMed]
- Jannesar, M.; Seyedi, S.M.; Jazi, M.M.; Niknam, V.; Ebrahimzadeh, H.; Botanga, C. A Genome-Wide Identification, Characterization and Functional Analysis of Salt-Related Long Non-Coding RNAs in Non-Model Plant Pistacia vera L. Using Transcriptome High Throughput Sequencing. Sci. Rep. 2020, 10, 5585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Ding, Z.; Tan, D.; Han, B.; Sun, X.; Zhang, J. Genome-Wide Discovery and Functional Prediction of Salt-Responsive LncRNAs in Duckweed. BMC Genom. 2020, 21, 212. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Zhang, Y.; Duan, M.; Huang, L.; Wang, W.; Xu, Q.; Yang, Y.; Yu, Y. Integrated Analysis of Long Non-Coding RNAs (LncRNAs) and MRNAs Reveals the Regulatory Role of LncRNAs Associated with Salt Resistance in Camellia sinensis. Front. Plant Sci. 2020, 11, 218. [Google Scholar] [CrossRef] [PubMed]
- Kotula, L.; Garcia Caparros, P.; Zörb, C.; Colmer, T.D.; Flowers, T.J. Improving Crop Salt Tolerance Using Transgenic Approaches: An Update and Physiological Analysis. Plant Cell Environ. 2020, 43, 2932–2956. [Google Scholar] [CrossRef] [PubMed]
- Segami, S.; Asaoka, M.; Kinoshita, S.; Fukuda, M.; Nakanishi, Y.; Maeshima, M. Biochemical, Structural and Physiological Characteristics of Vacuolar H+-Pyrophosphatase. Plant Cell Physiol. 2018, 59, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Feng, X.; Wang, L.; Su, Y.; Chu, Z.; Sun, Y. The Structure, Functional Evolution, and Evolutionary Trajectories of the H+-PPase Gene Family in Plants. BMC Genom. 2020, 21, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Search Item | Numbers |
|---|---|
| Total number of sequences examined | 48,442 |
| Total size of examined sequences (bp) | 109,925,036 |
| Total number of identified SSRs | 37,261 |
| Number of SSR-containing sequences | 21,573 |
| Number of sequences containing more than 1 SSR | 9024 |
| Number of SSRs present in compound formation | 5280 |
| Mononucleotide | 21,885 |
| Dinucleotide | 9024 |
| Trinucleotide | 5553 |
| Tetranucleotide | 466 |
| Hexanucleotide | 209 |
| Pentanucleotide | 124 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Liu, Z.; Hu, A.; Wu, H.; Zhu, J.; Wang, F.; Cao, P.; Yang, X.; Zhang, H. Full-Length Transcriptome Analysis of the Halophyte Nitraria sibirica Pall. Genes 2022, 13, 661. https://doi.org/10.3390/genes13040661
Zhang H, Liu Z, Hu A, Wu H, Zhu J, Wang F, Cao P, Yang X, Zhang H. Full-Length Transcriptome Analysis of the Halophyte Nitraria sibirica Pall. Genes. 2022; 13(4):661. https://doi.org/10.3390/genes13040661
Chicago/Turabian StyleZhang, Huilong, Zhen Liu, Aishuang Hu, Haiwen Wu, Jianfeng Zhu, Fengzhi Wang, Pingping Cao, Xiuyan Yang, and Huaxin Zhang. 2022. "Full-Length Transcriptome Analysis of the Halophyte Nitraria sibirica Pall" Genes 13, no. 4: 661. https://doi.org/10.3390/genes13040661
APA StyleZhang, H., Liu, Z., Hu, A., Wu, H., Zhu, J., Wang, F., Cao, P., Yang, X., & Zhang, H. (2022). Full-Length Transcriptome Analysis of the Halophyte Nitraria sibirica Pall. Genes, 13(4), 661. https://doi.org/10.3390/genes13040661