
A Novel Splice-Site Deletion in the POU1F1 Gene Causes Combined Pituitary Hormone Deficiency in Multiple Sudanese Pedigrees

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Anthropometry and Bone Age Estimation
2.2. Hormonal Testing
2.3. Magnetic Resonance Imaging (MRI)
2.4. Genetic Analysis of The POU1F1 Gene
2.5. Whole-Exome Sequencing (WES)
3. Results
3.1. The Phenotype of Patients with a Novel Variant in the POU1F1 Gene
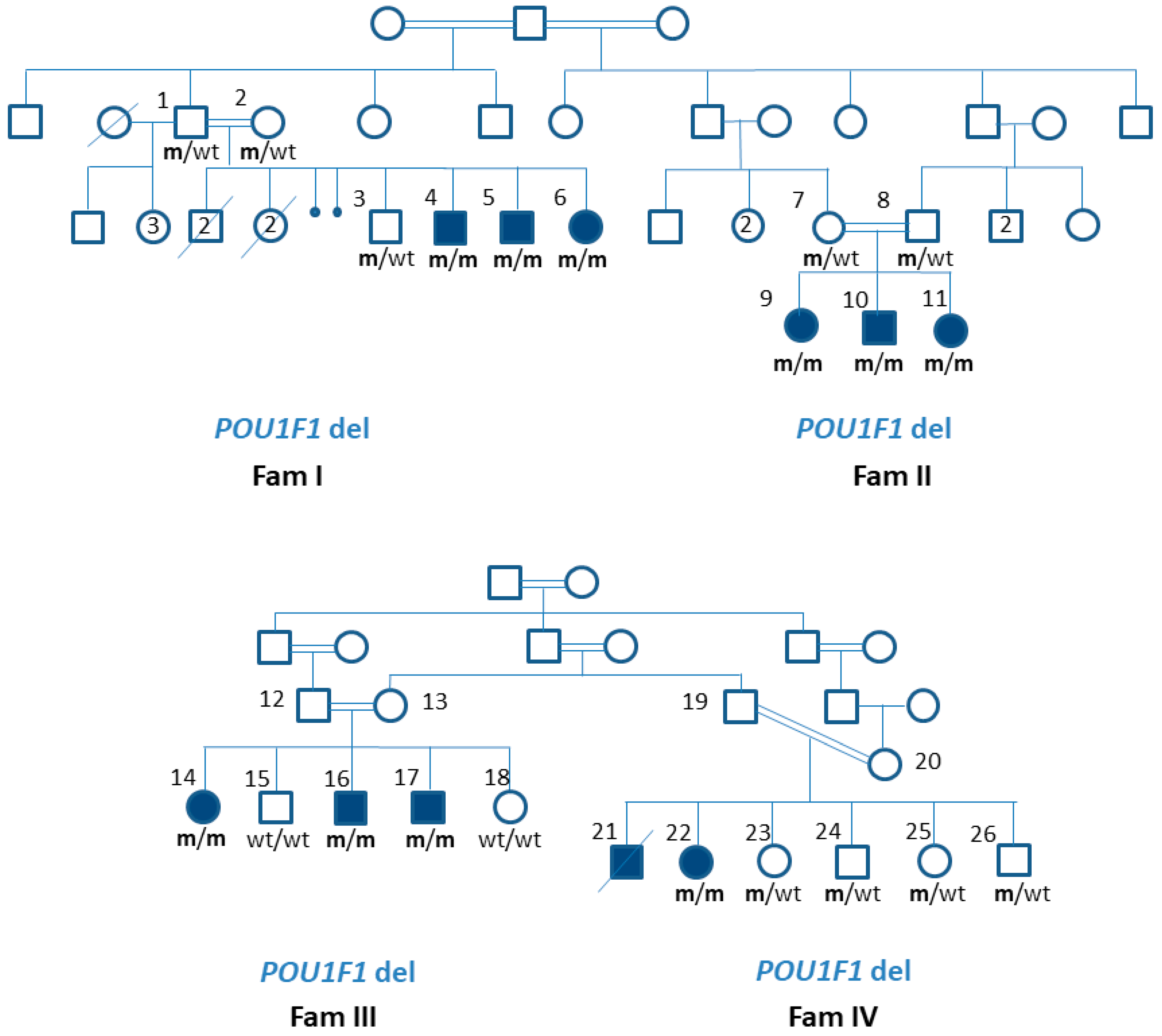
3.2. Patients’ Genotype Details
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gregory, L.C.; Dattani, M.T. The molecular basis of congenital hypopituitarism and related disorders. J. Clin. Endocrinol. Metab. 2020, 105, e2103–e2120. [Google Scholar] [CrossRef] [PubMed]
- Dattani, M.T. Novel insights into the aetiology and pathogenesis of hypopituitarism. Horm. Res. 2004, 62, 1–13. [Google Scholar] [CrossRef] [PubMed]
- De Rienzo, F.; Mellone, S.; Bellone, S.; Babu, D.; Fusco, I.; Prodam, F.; Petri, A.; Muniswamy, R.; De Luca, F.; Salerno, M.; et al. Frequency of genetic defects in combined pituitary hormone deficiency: A systematic review and analysis of a multicentre Italian cohort. Clin. Endocrinol. 2015, 83, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Blum, W.F.; Klammt, J.; Amselem, S.; Pfaffle, H.M.; Legendre, M.; Sobrier, M.L.; Luton, M.P.; Child, C.J.; Jones, C.; Zimmermann, A.G.; et al. Screening a large pediatric cohort with GH deficiency for mutations in genes regulating pituitary development and GH secretion: Frequencies, phenotypes and growth outcomes. EBioMedicine 2018, 36, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Turton, J.P.; Reynaud, R.; Mehta, A.; Torpiano, J.; Saveanu, A.; Woods, K.S.; Tiulpakov, A.; Zdravkovic, V.; Hamilton, J.; Attard-Montalto, S.; et al. Novel mutations within the POU1F1 gene associated with variable combined pituitary hormone deficiency. J. Clin. Endocrinol. Metab. 2005, 90, 4762–4770. [Google Scholar] [CrossRef] [PubMed]
- Ingraham, H.A.; Albert, V.R.; Chen, R.P.; Crenshaw, E.B., III; Elsholtz, H.P.; He, X.; Kapiloff, M.S.; Mangalam, H.J.; Swanson, L.W.; Treacy, M.N.; et al. A family of POU-domain and Pit-1 tissue-specific transcription factors in pituitary and neuroendocrine development. Annu. Rev. Physiol. 1990, 52, 773–791. [Google Scholar] [CrossRef]
- Simmons, D.M.; Voss, J.W.; Ingraham, H.A.; Holloway, J.M.; Broide, R.S.; Rosenfeld, M.G.; Swanson, L.W. Pituitary cell phenotypes involve cell-specific Pit-1 mRNA translation and synergistic interactions with other classes of transcription factors. Genes Dev. 1990, 4, 695–711. [Google Scholar] [CrossRef]
- Tatsumi, K.I.; Miyai, K.; Notomi, T.; Kaibe, K.; Amino, N.; Mizuno, Y.; Kohno, H. Cretinism with combined hormone deficiency caused by a mutation in the PIT1 gene. Nat. Genet. 1992, 1, 56–58. [Google Scholar] [CrossRef]
- Radovick, S.; Nations, M.; Du, Y.; Berg, L.A.; Weintraub, B.D.; Wondisford, F.E. A mutation in the POU-homeodomain of Pit-1 responsible for combined pituitary hormone deficiency. Science 1992, 257, 1115–1118. [Google Scholar] [CrossRef]
- Pfaffle, R.W.; DiMattia, G.E.; Parks, J.S.; Brown, M.R.; Wit, J.M.; Jansen, M.; Van der Nat, H.; Van den Brande, J.L.; Rosenfeld, M.G.; Ingraham, H.A. Mutation of the POU-specific domain of Pit-1 and hypopituitarism without pituitary hypoplasia. Science 1992, 257, 1118–1121. [Google Scholar] [CrossRef]
- Ohta, K.; Nobukuni, Y.; Mitsubuchi, H.; Fujimoto, S.; Matsuo, N.; Inagaki, H.; Endo, F.; Matsuda, I. Mutations in the Pit-1 gene in children with combined pituitary hormone deficiency. Biochem. Biophys. Res. Commun. 1992, 189, 851–855. [Google Scholar] [CrossRef]
- Jacobson, E.M.; Li, P.; Leon-del-Rio, A.; Rosenfeld, M.G.; Aggarwal, A.K. Structure of Pit-1 POU domain bound to DNA as a dimer: Unexpected arrangement and flexibility. Genes Dev. 1997, 11, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Alatzoglou, K.S.; Gregory, L.C.; Dattani, M.T. Development of the Pituitary Gland. Compr. Physiol. 2020, 10, 389–413. [Google Scholar] [CrossRef] [PubMed]
- Chapman, B.; Kirchner, R.; Pantano, L.; Naumenko, S.; Smet, M.D.; Beltrame, L.; Khotiainsteva, T.; Sytchev, I.; Guimera, R.V.; Kern, J.; et al. bcbio/bcbio-nextgen: (v.1.2.8). Zenodo 2021. [Google Scholar] [CrossRef]
- Desvignes, J.P.; Bartoli, M.; Delague, V.; Krahn, M.; Miltgen, M.; Beroud, C.; Salgado, D. VarAFT: A variant annotation and filtration system for human next generation sequencing data. Nucleic Acids Res. 2018, 46, W545–W553. [Google Scholar] [CrossRef]
- Abdelrahiem, S.K.; Bilal, J.A.; Al Nafeesah, A.; Al-Wutayd, O.; Rayis, D.A.; Adam, I. Low and high birth weight in a hospital population in Sudan: An analysis of clinical cut-off values. Int. J. Gynaecol. Obstet. 2021, 154, 427–430. [Google Scholar] [CrossRef]
- The Genome Aggregation Database. Available online: https://gnomad.broadinstitute.org (accessed on 10 February 2022).
- Salemi, S.; Besson, A.; Eble, A.; Gallati, S.; Pfaffle, R.W.; Mullis, P.E. New N-terminal located mutation (Q4ter) within the POU1F1-gene (PIT-1) causes recessive combined pituitary hormone deficiency and variable phenotype. Growth Horm. IGF Res. 2003, 13, 264–268. [Google Scholar] [CrossRef]
- Lee, N.C.; Tsai, W.Y.; Peng, S.F.; Tung, Y.C.; Chien, Y.H.; Hwu, W.L. Congenital hypopituitarism due to POU1F1 gene mutation. J. Formos. Med. Assoc. 2011, 110, 58–61. [Google Scholar] [CrossRef][Green Version]
- Sobrier, M.L.; Tsai, Y.C.; Perez, C.; Leheup, B.; Bouceba, T.; Duquesnoy, P.; Copin, B.; Sizova, D.; Penzo, A.; Stanger, B.Z.; et al. Functional characterization of a human POU1F1 mutation associated with isolated growth hormone deficiency: A novel etiology for IGHD. Hum. Mol. Genet. 2016, 25, 472–483. [Google Scholar] [CrossRef]
- Turton, J.P.; Strom, M.; Langham, S.; Dattani, M.T.; Le Tissier, P. Two novel mutations in the POU1F1 gene generate null alleles through different mechanisms leading to combined pituitary hormone deficiency. Clin. Endocrinol. 2012, 76, 387–393. [Google Scholar] [CrossRef]
- Brown, M.R.; Parks, J.S.; Adess, M.E.; Rich, B.H.; Rosenthal, I.M.; Voss, T.C.; VanderHeyden, T.C.; Hurley, D.L. Central hypothyroidism reveals compound heterozygous mutations in the Pit-1 gene. Horm. Res. 1998, 49, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Bas, F.; Abali, Z.Y.; Toksoy, G.; Poyrazoglu, S.; Bundak, R.; Gulec, C.; Uyguner, Z.O.; Darendeliler, F. Precocious or early puberty in patients with combined pituitary hormone deficiency due to POU1F1 gene mutation: Case report and review of possible mechanisms. Hormones 2018, 17, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Birla, S.; Vijayakumar, P.; Sehgal, S.; Bhatnagar, S.; Pallavi, K.; Sharma, A. Characterization of a Novel POU1F1 Mutation Identified on Screening 160 Growth Hormone Deficiency Patients. Horm. Metab. Res. 2019, 51, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Birla, S.; Khadgawat, R.; Jyotsna, V.P.; Jain, V.; Garg, M.K.; Bhalla, A.S.; Sharma, A. Identification of Novel PROP1 and POU1F1 Mutations in Patients with Combined Pituitary Hormone Deficiency. Horm. Metab. Res. 2016, 48, 822–827. [Google Scholar] [CrossRef]
- Malvagia, S.; Poggi, G.M.; Pasquini, E.; Donati, M.A.; Pela, I.; Morrone, A.; Zammarchi, E. The de novo Q167K mutation in the POU1F1 gene leads to combined pituitary hormone deficiency in an Italian patient. Pediatr. Res. 2003, 54, 635–640. [Google Scholar] [CrossRef]
- Pernasetti, F.; Milner, R.D.; al Ashwal, A.A.; de Zegher, F.; Chavez, V.M.; Muller, M.; Martial, J.A. Pro239Ser: A novel recessive mutation of the Pit-1 gene in seven Middle Eastern children with growth hormone, prolactin, and thyrotropin deficiency. J. Clin. Endocrinol. Metab. 1998, 83, 2079–2083. [Google Scholar] [CrossRef][Green Version]
- Fofanova, O.V.; Takamura, N.; Kinoshita, E.; Yoshimoto, M.; Tsuji, Y.; Peterkova, V.A.; Evgrafov, O.V.; Dedov, I.I.; Goncharov, N.P.; Yamashita, S. Rarity of PIT1 involvement in children from Russia with combined pituitary hormone deficiency. Am. J. Med. Genet. 1998, 77, 360–365. [Google Scholar] [CrossRef]
- McLennan, K.; Jeske, Y.; Cotterill, A.; Cowley, D.; Penfold, J.; Jones, T.; Howard, N.; Thomsett, M.; Choong, C. Combined pituitary hormone deficiency in Australian children: Clinical and genetic correlates. Clin. Endocrinol. 2003, 58, 785–794. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Cisternino, M.; Cohen, L.E. A novel nonsense mutation in the Pit-1 gene: Evidence for a gene dosage effect. J. Clin. Endocrinol. Metab. 2003, 88, 1241–1247. [Google Scholar] [CrossRef][Green Version]
- Hendriks-Stegeman, B.I.; Augustijn, K.D.; Bakker, B.; Holthuizen, P.; van der Vliet, P.C.; Jansen, M. Combined pituitary hormone deficiency caused by compound heterozygosity for two novel mutations in the POU domain of the Pit1/POU1F1 gene. J. Clin. Endocrinol. Metab. 2001, 86, 1545–1550. [Google Scholar] [CrossRef][Green Version]
- Irie, Y.; Tatsumi, K.; Ogawa, M.; Kamijo, T.; Preeyasombat, C.; Suprasongsin, C.; Amino, N. A novel E250X mutation of the PIT1 gene in a patient with combined pituitary hormone deficiency. Endocr. J. 1995, 42, 351–354. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gat-Yablonski, G.; Klar, A.; Hirsch, D.; Eliakim, A.; Lazar, L.; Hurvitz, H.; Phillip, M. Three novel mutations in POU1F1 in Israeli patients with combined pituitary hormone deficiency. J. Pediatr. Endocrinol. Metab. 2005, 18, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Blankenstein, O.; Muhlenberg, R.; Kim, C.; Wuller, S.; Pfaffle, R.; Heimann, G. A new C-terminal located mutation (V272ter) in the PIT-1 gene manifesting with severe congenital hypothyroidism. Possible functionality of the PIT-1 C-terminus. Horm. Res. 2001, 56, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.E.; Zanger, K.; Brue, T.; Wondisford, F.E.; Radovick, S. Defective retinoic acid regulation of the Pit-1 gene enhancer: A novel mechanism of combined pituitary hormone deficiency. Mol. Endocrinol. 1999, 13, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Gat-Yablonski, G.; Lazar, L.; Pertzelan, A.; Phillip, M. A novel mutation in PIT-1: Phenotypic variability in familial combined pituitary hormone deficiencies. J. Pediatr. Endocrinol. Metab. 2002, 15, 325–330. [Google Scholar] [CrossRef]
- Rainbow, L.A.; Rees, S.A.; Shaikh, M.G.; Shaw, N.J.; Cole, T.; Barrett, T.G.; Kirk, J.M. Mutation analysis of POUF-1, PROP-1 and HESX-1 show low frequency of mutations in children with sporadic forms of combined pituitary hormone deficiency and septo-optic dysplasia. Clin. Endocrinol. 2005, 62, 163–168. [Google Scholar] [CrossRef]
- Carlomagno, Y.; Salerno, M.; Vivenza, D.; Capalbo, D.; Godi, M.; Mellone, S.; Tiradani, L.; Corneli, G.; Momigliano-Richiardi, P.; Bona, G.; et al. A novel recessive splicing mutation in the POU1F1 gene causing combined pituitary hormone deficiency. J. Endocrinol. Investig. 2009, 32, 653–658. [Google Scholar] [CrossRef]
- Snabboon, T.; Plengpanich, W.; Buranasupkajorn, P.; Khwanjaipanich, R.; Vasinanukorn, P.; Suwanwalaikorn, S.; Khovidhunkit, W.; Shotelersuk, V. A novel germline mutation, IVS4+1G>A, of the POU1F1 gene underlying combined pituitary hormone deficiency. Horm. Res. 2008, 69, 60–64. [Google Scholar] [CrossRef]
- Inoue, H.; Mukai, T.; Sakamoto, Y.; Kimura, C.; Kangawa, N.; Itakura, M.; Ogata, T.; Ito, Y.; Fujieda, K.; Japan Growth Genome, C. Identification of a novel mutation in the exon 2 splice donor site of the POU1F1/PIT-1 gene in Japanese identical twins with mild combined pituitary hormone deficiency. Clin. Endocrinol. 2012, 76, 78–87. [Google Scholar] [CrossRef]
- Takagi, M.; Kamasaki, H.; Yagi, H.; Fukuzawa, R.; Narumi, S.; Hasegawa, T. A novel heterozygous intronic mutation in POU1F1 is associated with combined pituitary hormone deficiency. Endocr. J. 2017, 64, 229–234. [Google Scholar] [CrossRef]
- Tenenbaum-Rakover, Y.; Sobrier, M.L.; Amselem, S. A novel POU1F1 mutation (p.Thr168IlefsX7) associated with an early and severe form of combined pituitary hormone deficiency: Functional analysis and follow-up from infancy to adulthood. Clin. Endocrinol. 2011, 75, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Gat-Yablonski, G.; Frumkin-Ben David, R.; Bar, M.; Potievsky, O.; Phillip, M.; Lazar, L. Homozygous microdeletion of the POU1F1, CHMP2B, and VGLL3 genes in chromosome 3--a novel syndrome. Am. J. Med. Genet. A 2011, 155A, 2242–2246. [Google Scholar] [CrossRef] [PubMed]
- Bertko, E.; Klammt, J.; Dusatkova, P.; Bahceci, M.; Gonc, N.; Ten Have, L.; Kandemir, N.; Mansmann, G.; Obermannova, B.; Oostdijk, W.; et al. Combined pituitary hormone deficiency due to gross deletions in the POU1F1 (PIT-1) and PROP1 genes. J. Hum. Genet. 2017, 62, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Bas, F.; Uyguner, Z.O.; Darendeliler, F.; Aycan, Z.; Cetinkaya, E.; Berberoglu, M.; Siklar, Z.; Ocal, G.; Darcan, S.; Goksen, D.; et al. Molecular analysis of PROP1, POU1F1, LHX3, and HESX1 in Turkish patients with combined pituitary hormone deficiency: A multicenter study. Endocrine 2015, 49, 479–491. [Google Scholar] [CrossRef]
- Pellegrini-Bouiller, I.; Belicar, P.; Barlier, A.; Gunz, G.; Charvet, J.P.; Jaquet, P.; Brue, T.; Vialettes, B.; Enjalbert, A. A new mutation of the gene encoding the transcription factor Pit-1 is responsible for combined pituitary hormone deficiency. J. Clin. Endocrinol. Metab. 1996, 81, 2790–2796. [Google Scholar] [CrossRef][Green Version]
- Majdoub, H.; Amselem, S.; Legendre, M.; Rath, S.; Bercovich, D.; Tenenbaum-Rakover, Y. Extreme Short Stature and Severe Neurological Impairment in a 17-Year-Old Male With Untreated Combined Pituitary Hormone Deficiency Due to POU1F1 Mutation. Front. Endocrinol. 2019, 10, 381. [Google Scholar] [CrossRef]
- Chinyanga, E.; Chidede, O.; Mujaji, W.B. Thyroid function in neonates from goitre prevalent areas in Zimbabwe. Cent. Afr. J. Med. 1998, 44, 127–130. [Google Scholar]
- Das, S.C.; Isichei, U.P. A comparative study of thyroid function in African neonates: A reference thyroid profile. Clin. Chim. Acta 1993, 220, 233–238. [Google Scholar] [CrossRef]
- Bernstein, R.E.; Op’t Hof, J.; Hitzeroth, H.W. Neonatal screening for congenital hypothyroidism. A decade’s review, including South Africa. S. Afr. Med. J. 1988, 73, 339–343. [Google Scholar]
- Feleke, Y.; Enquoselassie, F.; Deneke, F.; Abdulkadir, J.; Hawariat, G.W.; Tilahun, M.; Mekbib, T. Neonatal congenital hypothyroidism screening in Addis Ababa, Ethiopia. East Afr. Med. J. 2000, 77, 377–381. [Google Scholar]
- Lauffer, P.; Zwaveling-Soonawala, N.; Naafs, J.C.; Boelen, A.; van Trotsenburg, A.S.P. Diagnosis and Management of Central Congenital Hypothyroidism. Front. Endocrinol. 2021, 12, 686317. [Google Scholar] [CrossRef] [PubMed]
- Naafs, J.C.; Verkerk, P.H.; Fliers, E.; van Trotsenburg, A.S.P.; Zwaveling-Soonawala, N. Clinical and genetic characteristics of Dutch children with central congenital hypothyroidism, early detected by neonatal screening. Eur. J. Endocrinol. 2020, 183, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Eckert-Lind, C.; Busch, A.S.; Petersen, J.H.; Biro, F.M.; Butler, G.; Brauner, E.V.; Juul, A. Worldwide Secular Trends in Age at Pubertal Onset Assessed by Breast Development Among Girls: A Systematic Review and Meta-analysis. JAMA Pediatr. 2020, 174, e195881. [Google Scholar] [CrossRef] [PubMed]
- Gillett-Netting, R.; Meloy, M.; Campbell, B.C. Catch-up reproductive maturation in rural Tonga girls, Zambia? Am. J. Hum. Biol. 2004, 16, 658–669. [Google Scholar] [CrossRef]
- Dasen, J.S.; O’Connell, M.; Flynn, S.E.; Treier, M.; Gleiberman, A.S.; Szeto, D.P.; Hooshmand, F.; Aggarwal, A.K.; Rosenfeld, M.G. Reciprocal interactions of Pit1 and GATA2 mediate signaling gradient-induced determination of pituitary cell types. Cell 1999, 97, 587–598. [Google Scholar] [CrossRef]
- Unal, O.; Tombul, T.; Cirak, B.; Anlar, O.; Incesu, L.; Kayan, M. Left hemisphere and male sex dominance of cerebral hemiatrophy (Dyke-Davidoff-Masson Syndrome). Clin. Imaging 2004, 28, 163–165. [Google Scholar] [CrossRef]
- Zuluaga, A.; Vargas, S.; Arango, S.; Uribe, R.U. Brain Asymmetry: Diagnostic Approach. Rev. Colomb. Radiol. 2017, 28, 7. [Google Scholar]
- Siegel, J.L. Acute bacterial meningitis and stroke. Neurol. Neurochir. Pol. 2019, 53, 242–250. [Google Scholar] [CrossRef]
- Zainel, A.; Mitchell, H.; Sadarangani, M. Bacterial Meningitis in Children: Neurological Complications, Associated Risk Factors, and Prevention. Microorganisms 2021, 9, 535. [Google Scholar] [CrossRef]
- Salvagno, G.L.; Pavan, C.; Lippi, G. Rare thrombophilic conditions. Ann. Transl. Med. 2018, 6, 342. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Family | Gender | Birth Weight (g) | Age at Diagnosis of Hypothyroidism (Days) | Age at Diagnosis of GHD (Years) | Height at Diagnosis of GHD (cm) (SD) | Bone Age in Years | Age at Start of Puberty (Years)/Menarche (m) | Mid Parenteral Height (cm) | End Adult Height (cm) | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|
| FI/4 | M | 3000 | 9 | 10 | 123 (−2.5) | NA | 15 | 172 | 153 | Short adult height |
| FI/5 | M | 2800 | 10 | 7 | 103 (−3.5) | 5 | 14 | 172 | 160 | Short adult height |
| FI/6 | F | 3100 | 4 | 5 | 100 (−1.7) | 4 | 13/MC = 16 | 159 | 144.5 | Short adult height |
| FII/9 | F | 2800 | 90 | 10 | 106 (−5.6) | 5.9 | 13/MC = 16 | 161 | 134 | Short adult height |
| FII/10 | M | 3000 | 60 | 4 | 86 (−4.6) | 1.7 | 15 | 174 | Still on GH | Still on GH |
| FII/11 | F | 3000 | 32 | 2 | 74 (−3.4) | 0.5 | 14/MC = 16 | 161 | Still on GH | Still on GH |
| FIII/14 | F | 2500 | 38 | 8 | 103 (−5.2) | 5 | 7/MC = 9.2 | 160.5 | 149 | Short adult height |
| FIII/16 | M | 2000 | 20 | 4 | 97 (−1.2) | NA | 14 | 173.5 | 165 | Reached genetic target range |
| FIII/17 | M | 2000 | 10 | 1.5 | 67 (−4.8) | NA | 13 | 173.5 | Still growing | Still on GH |
| FIV/21 | M | 3000 | 10 | 10 | 93.5 (−7.5) | NA | NA | 177 | Deceased | Deceased |
| FIV/22 | F | 2800 | 40 | 4 | 85 (−3.85) | 3 | 13/MC = 16 | 164 | 113 | Short adult height |
| Mean for parameters | 2450 | 29 | 5 | M = −4.01 F = −2.9 | - | M = 14.2 F = 12/MC = 14.6 | - |
| Family Number | Peak GH ng/mL (≥10) | Peak Cortisol µg/dL (≥18) | Am Cortisol µg/dL | TSH mIU/mL (0.2–4.9) | FT4 ng/dL | Prolactin ng/mL | Basal LH mIU/mL (≥0.3) |
|---|---|---|---|---|---|---|---|
| FI/4 | ˂0.07 | 22.4 | 14.7 | <0.02 | 1.2 (1.1–1.5) | 0.9 | 5.9 |
| FI/5 | ˂0.07 | 18.1 | 7.3 | 2.4 | 0.9 (1.1–1.6) | 0.9 | 2.7 |
| FI/6 | ˂0.07 | 16.1 | 14.8 | <0.02 | 1.5 (1.1–1.35) | 0.9 | 4.1 |
| FII/9 | ˂0.07 | 34.5 | NA | <0.01 | 4.1 (4.9–11) | 0.6 | 1.7 |
| FII/10 | N/A | 21.2 | NA | <0.005 | 3.2 (5.1–14.1) | 0.8 | 2.5 |
| FII/11 | N/A | 19.9 | NA | <0.005 | 7.2 (6.5–13.3) | 0.7 | 1.9 |
| FIII/14 | 2.1 | 13 | 15.7 | <0.02 | 2 (7.0–17) | ˂1.0 | 1.13 |
| FIII/16 | 2.1 | 15.5 | NA | 0.1 | 1.7 (7.0–17.0) | ˂1.0 | NA |
| FIII/17 | 0.05 | 24.9 | 11.4 | 0.3 | 6.6 (3.8–11.2) | NA | NA |
| FIV/21 | 0.31 | NA | 7 | <0.02 | NA | 0.1 | NA |
| FIV/22 | 0.36 | NA | 15.6 | <0.02 | 2.1 (2.9–5.1) | 0.1 | 22.1 |
| Family Number | MRI Findings |
|---|---|
| FI/4 | Hypoplastic anterior pituitary gland, normal posterior gland, and intact stalk |
| FI/5 | Hypoplastic anterior gland, normal posterior gland, and intact stalk |
| FI/6 | NA |
| FII/9 | Empty sella turcica |
| FII/10 | NA |
| FII/11 | NA |
| FIII/14 | Large left cerebral infarction with normal pituitary gland |
| FIII/16 | NA |
| FIII/17 | Hypoplastic anterior gland |
| FIV/21 | Unilateral cerebral diffuse cortical gliosis, atrophy with evacuee dilatation of the ipsilateral ventricle, prominent sulci, and hypoplastic anterior pituitary gland |
| FIV/22 | Right cerebral hemiatrophy, with marked loss of white, subcortical, and deep matter volumes with prominent sulci, and secondary ballooning of the right lateral ventricle with no midline shifting, right temporal cystic encephalomalacia |
| Index Case | Blood Glucose mg/dL (mmo/L) | Cortisol ng/mL (mmol/L) |
|---|---|---|
| FIII/14 | 32 (1.8) | 12.1 (333.8) |
| FIII/16 | 36 (2.0) | 6.1 (168.2) |
| FIII/17 | 45 (2.5) | 10.7 (295.2) |
| FIV/22 | 40 (2.2) | 7.0 (193.1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, S.S.; Abdullah, M.; Trebusak Podkrajsek, K.; Musa, S.; Ibrahim, A.; Babiker, O.; Kovac, J.; Battelino, T.; Avbelj Stefanija, M. A Novel Splice-Site Deletion in the POU1F1 Gene Causes Combined Pituitary Hormone Deficiency in Multiple Sudanese Pedigrees. Genes 2022, 13, 657. https://doi.org/10.3390/genes13040657
Hassan SS, Abdullah M, Trebusak Podkrajsek K, Musa S, Ibrahim A, Babiker O, Kovac J, Battelino T, Avbelj Stefanija M. A Novel Splice-Site Deletion in the POU1F1 Gene Causes Combined Pituitary Hormone Deficiency in Multiple Sudanese Pedigrees. Genes. 2022; 13(4):657. https://doi.org/10.3390/genes13040657
Chicago/Turabian StyleHassan, Samar S., Mohamed Abdullah, Katarina Trebusak Podkrajsek, Salwa Musa, Areej Ibrahim, Omer Babiker, Jernej Kovac, Tadej Battelino, and Magdalena Avbelj Stefanija. 2022. "A Novel Splice-Site Deletion in the POU1F1 Gene Causes Combined Pituitary Hormone Deficiency in Multiple Sudanese Pedigrees" Genes 13, no. 4: 657. https://doi.org/10.3390/genes13040657
APA StyleHassan, S. S., Abdullah, M., Trebusak Podkrajsek, K., Musa, S., Ibrahim, A., Babiker, O., Kovac, J., Battelino, T., & Avbelj Stefanija, M. (2022). A Novel Splice-Site Deletion in the POU1F1 Gene Causes Combined Pituitary Hormone Deficiency in Multiple Sudanese Pedigrees. Genes, 13(4), 657. https://doi.org/10.3390/genes13040657