Abstract
Pediatric high-grade gliomas (pHGGs) encompass a heterogeneous group of tumors. Three main molecular types (H3.3 mutant, IDH mutant, and H3.3/IDH wild-type) and a number of subtypes have been identified. We provide an overview of pHGGs and present a mono-institutional series. We studied eleven non-related pHGG samples through a combined approach of routine diagnostic tools and a gene panel. TP53 and H3F3A were the most mutated genes (six patients each, 54%). The third most mutated gene was EGFR (three patients, 27%), followed by PDGFRA and PTEN (two patients each, 18%). Variants in the EZHIP, MSH2, IDH1, IDH2, TERT, HRAS, NF1, BRAF, ATRX, and PIK3CA genes were relatively infrequent (one patient each, 9%). In one case, gene panel analysis documented the presence of a pathogenic IDH2 variant (c.419G>A, p.Arg140Gln) never described in gliomas. More than one-third of patients carry a variant in a gene associated with tumor-predisposing syndromes. The absence of constitutional DNA did not allow us to identify their constitutional origin.
Keywords:
pediatric; high-grade glioma; astrocytoma; thalamic glioma; gene panel; IDH2 mutation; EZHIP; TP53; H3F3A; case report 1. Introduction
Central nervous system (CNS) tumors are the most common solid neoplasms in childhood [1]. Among these, approximately 50% are gliomas [1]. Unlike in adults, low-grade gliomas (LGGs) predominate in children, while high-grade gliomas (HGGs) are less frequent [2,3]. The incidence of pediatric HGGs (pHGGs) has been calculated as 1.1–1.78 per 100,000 children [4]. Despite their low incidence, pHGGs are responsible for over 40% of all childhood brain tumor death, and overall, they are the more common cause of tumor-related death at this age [1,3]. Although the morphological features of pHGGs are comparable to those of HGGs in adulthood, the genetic–molecular characteristics differ so much that some of the novel therapeutic strategies derived from research on adult gliomas have yielded unsatisfactory results [5]. In addition, while adult HGGs are more often restricted to the cerebral hemispheres, those affecting children can occur throughout the central nervous system, with about half of cases occurring in midline locations [4]. Important molecular differences also exist within pHGGs. In this regard, on the basis of the genomic, epigenomic and transcriptomic profile, three main molecular types and a series of molecular subtypes—each with clinical peculiarities (i.e., age, tumor location, prognosis, and potential targetable therapy)—have been identified in the current scientific publications [4,5,6]. The three main molecular types of pHGGs are the histone H3 mutant, the isocitrate dehydrogenase gene (IDH) mutant, and the H3/IDH wild-type [4,5,6]. Additionally, there are the infant-type hemispheric gliomas and diffuse-midline glioma epidermal growth factor receptor (EGFR) mutant [7,8]. The molecular stratification of pHGGs into types and subtypes is essential due to the availability of promising new targeted therapies.
1.1. Histone H3 Mutant pHGGs
About 40% of pHGGs fall into the subgroup in which the genes encoding for histone H3 are mutated [5]. H3 is one of the major families of histones. Histones play an essential role in the condensation of DNA by contributing to the formation of nucleosomes. They may undergo post-translational modifications (i.e., methylation, acetylation and phosphorylation) that modify their interaction with DNA and alter a number of biological processes such as gene expression, DNA repair, mitosis and meiosis [9].
The majority of H3 mutant pHGGs harbor variants at position 27 consisting of a substitution of lysine with methionine (Lys27Met) in the H3.3 (H3F3A) gene (about three-fourths) or in H3.1 (HIST1H3B/C) gene (about one-fourth). The consequence of these variants is the biochemical inhibition of Polycomb Repressor Complex 2 (PRC2) (a protein exhibiting histone methyltransferase activity) through the sequestration of its catalytic subunit Enhancer of Zeste Homolog 2 (EZH2). The result is a global loss of trimethylation of lysine 27 (H3.3K27me3) on all H3 molecules both mutated and wild type [10]. The inhibition of the methylation pathways promotes tumorigenesis through several modalities, particularly by altering of the dynamics of chromatin. H3 mutant tumors typically arise in the midline (pons, midbrain, thalamus, spinal cord) and usually affect school-aged children. H3.3 mutant HGGs have a very dismal prognosis despite the fact that the histological features may sometimes be consistent with LGGs. H3.1 mutant HGGs seem to respond slightly better to radiotherapy with a consequent less aggressive course [11]. It is debated whether the dismal prognosis of Lys27Met mutated gliomas is related to the midline location—which prevents surgical resection in most cases—or it is an independent prognostic factor. Indeed, a number of low-grade and non-midline tumors carry this variant without influencing prognosis [12,13].
H3F3A may also be mutated at position Gly34 due to the substitution of glycine with arginine or valine (H3.3G34R/V). Gly34Arg/Val results in a total reduction of H3K36me2 and H3K36me3 levels [9,14]. Moreover, Gly34Val affects the regulation of gene expression and results in up-regulation of MYCN [15]. An additional molecular feature of H3G34 mutant gliomas is the high frequency of O6-methylguanine DNA methyltransferase gene (MGMT) promoter methylation. Tumor-harboring Gly34Arg/Val variants are typically hemispheric and, when compared with H3.3K27M mutated gliomas, affect older age group (teenagers and young adults); they are associated with a slightly prolonged survival, possibly related to MGMT methylation, resulting in enhanced responsiveness to temozolomide [1,16,17].
1.2. IDH Mutant pHGGs
IDH genes, which encode for IDH enzymes, are frequently mutated in LGGs and secondary glioblastomas. IDH proteins are involved in the Krebs cycle, catalyzing the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG). Variants in IDH inhibit the physiologic activity of the IDH proteins in converting isocitrate to α-KG, instead determining its conversion to 2-hydrosxyglutarate (2-HG) [18]. All variants result in an approximately 100-fold increase of 2-HG which is an oncometabolite that contributes to tumor development. Moreover, the IDH mutated proteins upregulate vascular endothelial growth factor (VEGF) and result in high levels of hypoxia-inducible factor-1α (HIF-1α), which can both ultimately contribute to glioma genesis [18]. Among the IDH family genes, IDH1 and IDH2 are the most frequently mutated in gliomas. Point variants of IDH1 are by far the most frequent. IDH1 and IDH2 variants are heterozygous, of somatic origin, and mutually exclusive (cases of concurrent IDH1 and IDH2 variants are exceedingly rare) [19]. IDH1 variants are strongly associated with astrocytomas TP53 and alpha thalassemia/mental retardation syndrome X-linked (ATRX) mutated, while IDH2 variants predominantly occur in oligodendrogliomas ATRX wild type and 1p-19q codeleted. Approximately 90% of IDH1 variants are located in exon 4 at codon 132 where, in the majority of the cases, a CGT–CAT transition changes a single amino acid from arginine to histidine (Arg132His). In the few remaining cases, different variants have been described but are usually restricted to codon 132, which is the isocitrate-binding pocket of IDH1. IDH2 variants are exclusively detected in arginine at position 172, which is the analogous site to arginine 132 in IDH1. Both low- and high-grade gliomas may harbor IDH variants. However, when cyclin-dependent kinase inhibitor 2A/B (CDKN2A/B) homozygous deletion co-occurs, the highest grade (grade 4) is assigned [6]. IDH variants are typically observed in adulthood, whereas they are rare at pediatric age [19]. Contrary to adult cases, IDH-mutated pHGGs do not show evidence of lower-grade precursor lesions. IDH-mutant pHGGs typically affect cerebral hemispheres and, similarly to what it observed in adults, have more favorable behavior and are likely to be associated with MGMT methylation, making temozolomide a potentially effective treatment option. Many pediatric IDH-mutated gliomas do not harbor the Arg132His with high incidence of rare variants such as Arg132Gly, Arg132Ser and Arg132Cys [2,20,21].
1.3. H3/IDH Wild Type pHGGS
The third molecular type of pHGG corresponds to H3/IDH wild-type lesions. This type is highly heterogeneous, comprising a number of largely hemispheric tumors affecting infants/newborns, children and adolescents, and displaying a variety of genomic and epigenetic features as well as different clinical behaviors. In particular, in this third molecular type fall pleomorphic-xantoastrocytoma (PXA)-like pHGGs, v-myc myelocytomatosis viral-related oncogene (NMYC), platelet-derived growth factor receptor A (PDGFRA), or EGFR amplified gliomas and hypermutant pHGGs [5,22].
1.4. PXA-like pHGGs
PXA-like pHGGs are hemispheric lesions molecularly characterized by the v-raf murine sarcoma viral oncogene homolog B1 (BRAF) Val600Glu variant, usually along with CDKN2A/B gene deletion [6]. The BRAF gene is a proto-oncogene coding a protein also called BRAF. BRAF is a member of the rapidly accelerated fibrosarcoma (RAF) kinase family, which transduces signals downstream of the rat sarcoma viral oncogene homolog (RAS) via the mitogen-activated protein kinase (MAPK) pathway, playing a role in cell growth [23]. Many variants in the BRAF gene have been identified in cancer. These variants lead to extracellular signal-regulated kinase (ERK) activation. The val600Glu variant, consisting of the substitution of valine to glutamic acid at position 600, is the commonest. The CDKN2A/B tumor suppressor genes encode for p16 and p15 proteins. p16 and p15 inhibit cyclin-dependent kinase proteins, thus activating the retinoblastoma protein family with a consequent block of the transition from the G0-phase to the S-phase of the cell cycle. In CNS, both high-grade and low-grade tumors may carry Val600Glu and CDKN2A/B deletion. PXAs and gangliogliomas have the highest incidence of Val600Glu. PXA-like pHGGs have a better prognosis, even though they exhibit a high rate of recurrence [5,23,24].
1.5. pHGGs Amplified in NMYC, PDGFRA or EGFR
pHGGs amplified in NMYC, PDGFRA and EGFR may arise in children of any age and represent distinct molecular sub-types associated with significantly different behavior: MYCN-amplified gliomas show the poorest prognosis; PDGFRA-amplified gliomas, designated as RTK1 pHGGs, show a better prognosis; and the EGFR-amplified gliomas, designated RTK2 pHGGs, show an intermediate prognosis between the previous two [22].
MYCN is a member of the MYC family of oncogenes. It is highly expressed in neural tissue during embryogenesis. It encodes a basic helix-loop-helix–leucine zipper (bHLH-LZ) protein called N-Myc or MYCN. This protein plays a role in the regulation of gene transcription. The amplification of the MYCN gene is associated with a variety of tumors, most notably neuroblastoma, in which the level of amplification is related to a dismal prognosis. A small number of high-grade brain tumors, quantified in 8–9% of cases, may disclose MYCN amplification [22,25,26].
PDGFRA and EGFR encode for high-affinity cell surface receptors belonging to the receptor tyrosine kinase (RTKs) family. They are involved in important physiologic cellular processes. RTKs are generally activated by the interaction of receptor-specific RTK ligands. Alterations in genes encoding RTKs lead to interference in a series of signaling pathways involved in determining oncogenesis and neoplastic progression. PDGFRA is essential for glial development and is involved in gliomagenesis through different mechanisms such as PDGFRA amplification or activating variants [3,27]. Alterations in PDGF-driven signaling are prevalent in the majority of pediatric tumors, while EGF-driven signaling is predominant in adults [28,29].
1.6. Hyper Mutant pHGGs
Hyper-mutant pHGGs affect children of any age, are more often hemispheric, and are associated with a poor prognosis [5]. These tumors are characterized by microsatellite instability (MSI). Microsatellites are short, repeated segments of DNA (mononucleotide, dinucleotide, or higher-order nucleotide repeats). MSI mostly depends on the presence of inactivating variants in the DNA mismatch repair (MMR) genes, mainly mutL homologue 1 (MLH1), mutS homologue 2 (MSH2), mutS homologue 6 (MSH6), and postmeiotic segregation increased 2 (PMS2). The resulting accumulation of variants can lead to oncogenesis and neoplastic progression. MSI has been described in HGGs with a frequency ranging from a percentage close to 0% to more than 40%, probably as consequence of the different sensitivities of the method used to ascertain the MSI status and of the age of the patients studied [30]. In fact, MSI-related gliomas are more frequent in children than in adults. Variants in MMR genes may be sporadic or inherited as a monoallelic germline variant (Lynch syndrome) or biallelic variant (constitutional mismatch repair-deficiency, CMMRD) [31]. Moreover, MMR-acquired variants or overexpression correlate with temozolomide (TMZ) resistance in glioblastomas [32,33,34].
1.7. Infant-Type Hemispheric Gliomas
Infant-type hemispheric gliomas are rare pathological entities, clinically distinct from gliomas, that arise in older children. In fact, they are associated with a better outcome regardless of the histological grade (these tumors may encompass a wide range of histological features from low-grade to frankly high-grade morphology), even with incomplete surgical resection and the age-related impossibility of radiotherapy. The most common molecular alterations observed in HGGs occurring in infants are fusion events, particularly those involving neurotropic receptor tyrosine Kinase (NTRK)1/2/3. NTRK1/2/3 encodes for tropomyosin receptor kinases (Trk) A, B and C, respectively [7]. Trk proteins belong to a family of growth factor receptors. They regulate synaptic strength and plasticity in the mammalian nervous system. The fusion genes resulting from the juxtaposition of the C-terminal kinase domain of NTRK1/2/3, with the N-terminal sequences of different genes, lead to the transcription of chimeric Trk proteins with oncogenic potential. AGBL carboxypeptidase 4 (AGBL4)4:NTRK2, tropomyosin 3 (TPM3):NTRK1, and ETS variant transcription factor 6 (ETV6):NTRK3 fusions are the more frequent fusions observed in infantile HGGs. Other fusion genes in infants involve anaplastic lymphoma kinase (ALK), c-ros oncogene 1 (ROS), and mesenchymal-epithelial transition (MET); ALK is more often involved in LGGs, and ROS and MET in HGGs. The availability of anti-Trk drugs represents an interesting therapeutic opportunity for these patients [7].
1.8. Diffuse Midline Gliomas EGFR Mutant
Thalamic gliomas—mainly bilateral at the onset, showing EGFR variants, EZHIP (ex CXorf67) overexpression, and loss of H3.3K27me3 in the absence of H3F3A variants—have recently been described. They are rare and typically affect young children. Due to the impossibility of an effective surgical treatment, the prognosis is invariably poor irrespective of histological grade. Recent studies have documented that in the diffuse midline glioma EGFR mutant, PRC2 inhibition and the consequent H3.3K27me3 loss is related to an epigenetic aberrant overexpression of the EZH2 inhibitory protein EZHIP, instead of to H3F3A variants. The oncoprotein EZHIP and H3.3K27M are competitive inhibitors of PRC2 [35,36,37]. Considering that H3.3K27me3 loss is the main molecular feature of both the H3.3K27M mutant and diffuse midline EGFR mutant gliomas, it has been proposed that these tumors be grouped under the denomination of H3.3K27-altered pHGGs [36,38].
1.9. Aim
We present a mono-institutional series of pHGGs operated at the Surgical Unit of the Meyer Children’s Hospital of Florence. Our aim was to evaluate the presence of a number of further molecular alterations in addition to those evaluated for strictly diagnostic purposes.
2. Patients and Methods
2.1. Patients
Eleven non-related consecutive pHGGs for which fresh material was available for molecular study were used in the study. Six (55%) patients were female, and five (45%) were male. The mean age at diagnosis was 7 years (range 2 months–18 years). Five lesions were hemispheric (45%), two were thalamic (18%) and the remaining four tumors, respectively, arose from the midbrain, hypothalamus, cerebellum and spinal cord (9% for each one) (Table 1).

Table 1.
Clinical data and molecular classification. F: female; M: male.
2.2. Methods
The original slides were re-evaluated according to the latest guidelines of the World Health Organization (WHO) Classification of CNS tumors [38]. Moreover, in order to molecularly characterize the tumors, a combined approach of routine diagnostic tools (immunohistochemistry: formalin-fixed and paraffin-embedded samples, standard streptavidin-biotin technique and commercially available antibodies against H3.3K27me3 and p53; fluorescence in situ hybridization (FISH) for determining MYCN, EGFR, PDGFRA and 1p19q status: formalin-fixed and paraffin-embedded samples and commercially available dual color FISH probes; molecular evaluations: formalin fixed and paraffin embedded samples, automated RNA extraction and PCR real-time amplification of the NTRK genes fusion panel), and gene panel (see below) was used.
The different additional analyses for each case were also chosen considering the location of tumor and the age of the patient.
2.3. DNA Extraction
Tumor DNAs were extracted using QIAamp Mini Kit (QIAGEN®, Hilden, Germany), according to the manufacturer’s instructions, and quantified using a NanoDROP 2000 Spectrophotometer (Thermo Scientific, Waltham, MA, USA).
2.4. Gene Panel and Bioinformatics Analysis of pHGG Panel Genes
Tumor DNA libraries were constructed using an enzymatic strategy to produce dsDNA fragments, followed by end repair, A-tailing, adapter ligation, and library amplification (Kapa Biosystems, Wilmington, MA, USA). A library pool was hybridized with SeqCap EZ Exome v3 probes (Nimblegen, Roche, Basel, Switzerland), and sequenced using NextSeq550 (Illumina Inc., San Diego, CA, USA). The reads obtained were aligned with the human reference genome hg19 using a Burrows–Wheeler Aligner (BWA, Wellcome Trust Sanger Institute, Wellcome Genome Campus, Cambridge, CB10 1SA, UK), and mapped and analyzed with IGV (Integrative Genome Viewer, IGV Broad Institute and Regents of the University of California) software.
The variant call for the identification of nucleotide variants of 34 genes (AKT1, ALK, ATRX, BRAF, CDK4, CDK6, CDKN2A, CDKN2B, EGFR, EZH2, EZHIP, H3F3A, IDH1, IDH2, KDM6A, KRAS, HRAS, NRAS, MAP2K1, MAP2K2, MLH1, MLH3, MSH2, MSH6, NF1, NF2, NTRK1, PDGFRA, PIK3CD, PIK3CA, PTEN, ROS1, TERT, TP53), was performed using automated in-house pipelines.
The paired-end reads were aligned to the human hg19 reference genome sequence using the Burrows–Wheeler Aligner v0.7.10 (BWA; http://bio-bwa.sourceforge.net, 10 November 2021, and variant calling was performed using the UnifiedGenotyper module of the Genome Analysis ToolKit v3.3-0 (GATK; https://gatk.broadinstitute.org, 9 April 2021). To discover the somatic variants present in tumor tissue, variant calling was performed using the MuTect method [39]. The final variant calling format (VCF) files were filtered and annotated using ANNOVAR software [40]. Variants that were called less than 20X, were off-target, were synonymous, or had a minor allele frequency (MAF) >5% in the Exome Aggregation Consortium (ExAC), Cambridge, MA, USA (http://exac.broadinstitute.org, 9 April 2021) were eliminated.
All variants were confirmed using Sanger sequencing. All validated variants of selected target genes were classified as pathogenic, likely pathogenic, uncertain clinical significance (VUS), uncertain clinical significance LP (VUS with minor pathogenic evidence) or benign, in agreement with the interpretation guidelines of the American College of Medical Genetics and Genomics (ACMG) [41]. Their tumor or disease association were evaluated in the somatic (Catalogue of Somatic Mutations in Cancer, COSMIC, https://cancer.sanger.ac.uk/cosmic, 14 June 2021) and constitutional (Human Gene Mutation Database, HGMD, http://www.hgmd.cf.ac.uk, 14 June 2021) databases, to clarify their hypothetical pathogenetic roles.
In silico analysis determined the pathogenicity of the variants of the splice site (BGDP https://www.fruitfly.org/seq_tools/splice.html, 14 June 2021) and the effect of the variants on the protein function (fathmm-MKL, https://fathmm.biocompute.org.uk/fathmmMKL.htm, 14 June 2021; FATHMM, http://fathmm.biocompute.org.uk/, 14 June 2021; MutationTaster, https://www.mutationtaster.org/, 14 June 2021; MutationAssessor, http://mutationassessor.org/r3/, 14 June 2021; SIFT, https://sift.bii.a-star.edu.sg/, 14 June 2021, Polyphen2_HVAR, http://genetics.bwh.harvard.edu/pph2/, 14 June 2021).
3. Results
Three tumors (27%) (P2, P5 and P9) are classified in the H3.3K27M mutant sub-type, two (18%) (P10 and P11) in the H3.3G34R/V mutant sub-type, one (9%) (P3) in the IDH mutant sub-type (Figure 1), one (9%) (P6) as a PXA-like pHGG, and one (9%) (P4) as a diffuse midline glioma EGFR mutant (Figure 2).
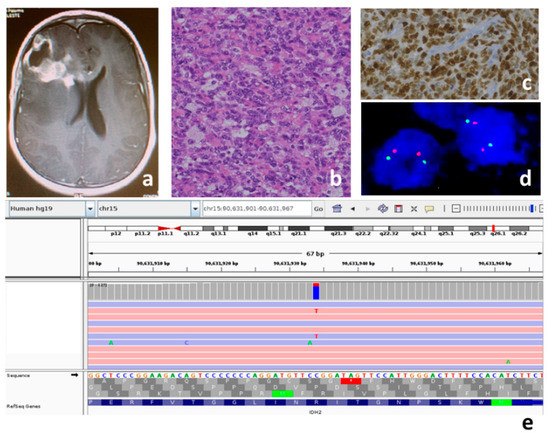
Figure 1.
Patient P3, IDH mutant pHGG: (a) MRI T1 contrast-weighted; right frontal lesion, unevenly capturing contrast, extensive periwound edema; the tumor is in contact with the frontal horn of the right lateral ventricle; (b) medium-sized atypical cells and intralesional histiocytes, hematoxylin and eosin, original magnification 20×; (c) positive nuclear immunostaining for p53 protein as indirect expression of TP53 mutation, original magnification 20×; (d) no 1p36 deletion in dual color FISH analysis (two orange and two red signals in each nucleus); (e) the variant c.419G>A p.Arg140Gln in exon 4 of the IDH2 gene (NM_002168) is read in both the strands 119X (94X in G ref allele and 23X in T alt allele).

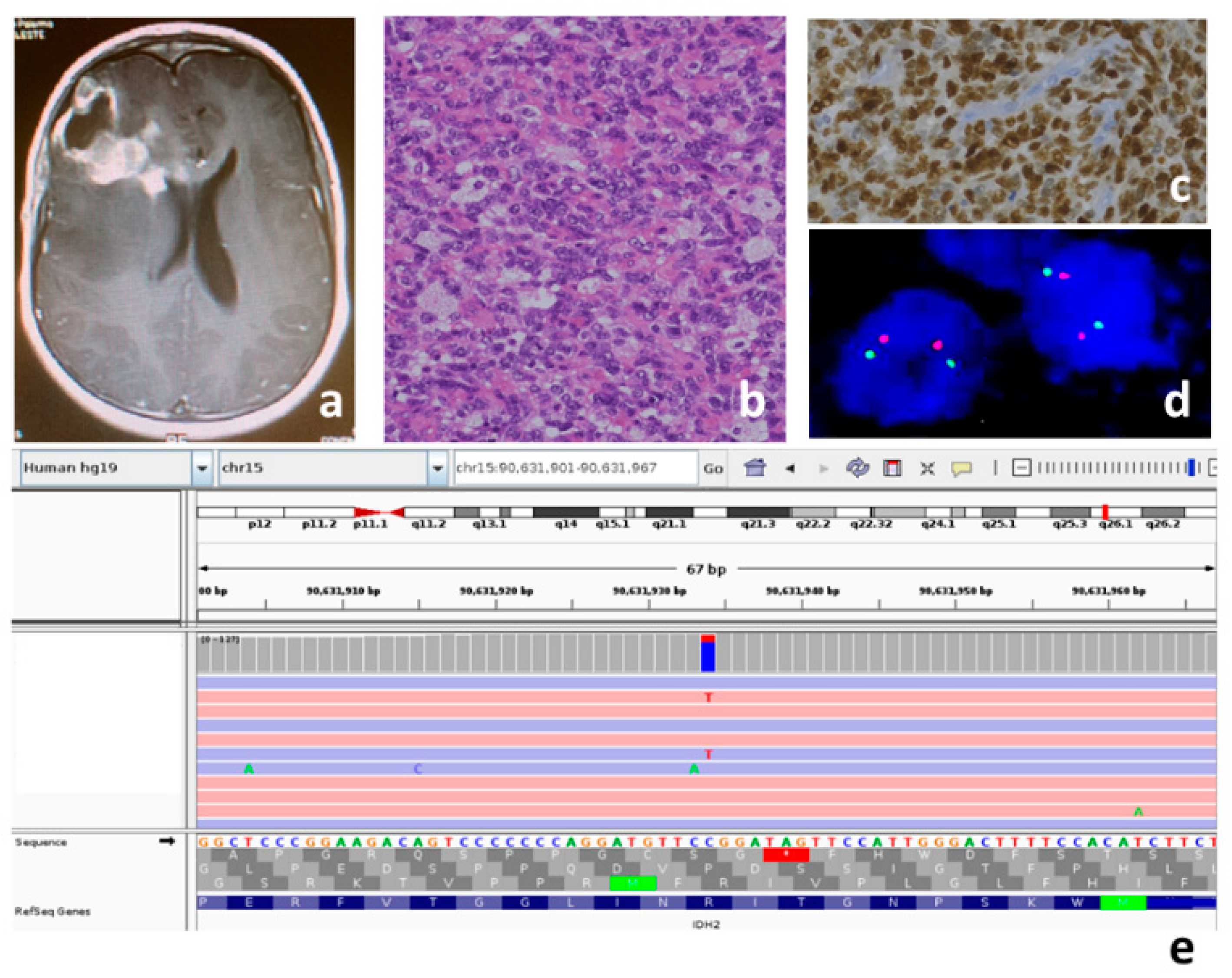
Figure 2.
Patient P4, thalamic diffuse EGFR mutant pHGG: (a) T2-weighted image, presence of hyperintense mass in the right thalamic area. The tumor is in contact with the medial portion of the third ventricle and the occipital horns. No development of hydrocephalus; (b) spindle-shaped atypical cells, hematoxylin and eosin, original magnification 20×; (c) negative nuclear staining for H3.3K27M-mutant protein, original magnification 20×; (d) negative nuclear staining for H3.3K27me3 in tumor cells and positive control in the endothelial cells, original magnification 20×; (e) the variant c.2300_2301insCAGCGTGGA p.Ala767_Ser768insAlaTrpThr in exon 20 of the EGFR gene (NM_005228) is read in both the strands 42X.
In the remaining three cases (36%) (P1, P7 and P8), the analyses carried out did not allow us to sub-classify them. (Table 1) In particular, immunohistochemistry and the successive gene panel excluded the H3F3A variants, the Val600Gln variant in the BRAF gene, and the MMR genes variants. FISH analyses executed to ascertain MYCN, EGFR and PDGFR amplification, and PDGFRA deletion, gave negative results for P1 and non-evaluable results for P7 and P8. For patient P1, considering the age and hemispheric localization of the tumor, we also ruled out the possibility of it being an infant-type pHGG through a search for fusions typical of this glioma. For patient P3, considering the presence of the IDH2 variant and the ATRX wild type (Table 2), the possible presence of 1p-19q codeletion was ruled out using FISH analysis (Figure 1, Table 1).
Table 2.
Variants identified by gene panel analysis. Uncertain Significance LP: VUS with minor pathogenic evidence; LOH: Loss of Heterozygosity; allele frequency from gnomAD (https://gnomad.broadinstitute.org/, 28 February 2022 NFE-population; *: HGVS (https://varnomen.hgvs.org/, 14 June 2021) stop codon.
The gene panel results are summarized in Table 2.
No variants were identified in the following target genes: AKT1, ALK, CDK4, CDK6, CDKN2A, CDKN2B, EZH2, KDM6A, KRAS, NRAS, MAP2K1, MAP2K2, MLH1, MLH3, MSH6, NF2, NTRK1, PIK3CD, ROS1.
All cases have at least one variant in the target genes, and most have 2–4 variants, with the exception of P3 and P11, which have nine and seven variants, respectively. Only P1 and P4 presented a single variant.
Our cases showed variants in TP53 (six patients, 54%), H3F3A (six patients, 54%), EGFR (three patients, 27%), PDGFRA, and phosphatase and tensin homolog (PTEN) genes (two patients each, 18%). Variants in the EZHIP, MSH2, IDH1/2, TERT, HRAS, NF1, BRAF, ATRX and Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha (PIK3CA) genes were less frequent (one patient each, 9%).
Five in six (83%) TP53 mutated tumors present a double inactivation of the TP53 gene (P2, P3, P8, P11 with double pathogenic variants and P10 with a pathogenic variant plus loss of heterozygosity (LOH)), while P9 shows only one hit. The TP53 variants are five missense and four premature stop codon/frameshift (50%, respectively), localized in the core (4–8 exons) (80%) of the gene. All variants are pathogenic.
H3F3A variants are recurrent at the Lys27 hotspot in three in six (50%) patients (P2, P5 and P9), and in the Gly34 hotspot in two in six (33%) patients (P10 and P11). c.83A>T (p.Lys27Met) is a pathogenic variant in exon 2; instead Gly34 presents two different amino acid substitutions (c.103G>A p.Gly34Arg in P10, and c.104G>T p.Gly34Val in P11). Only one in six (16%) patients (P3) has c.246T>G (p.Asp82Glu) in exon 3 of H3F3A gene.
One in three (33%) EGFR-mutated tumors presents multiple variants (P7). None of the EGFR variants are benign or are distributed over the entire gene (3–20 exons).
One in two (50%) PDGFRA-mutated tumors showed two variants (P3).
Two in two (100%) PTEN mutated tumors simultaneously carried a single variant (c.302delT and c.781C>G) and the LOH of the wild-type allele of the PTEN gene (P10 and P11).
Unique variants are also identified in unshared genes in several patients (P1, P3, P5, P6, P9, P10 and P11).
P1 presents an uncertain significance variant (c.1328C>T p.Ser448Phe) in exon 1 of the EZHIP gene. P3 presents an uncertain significance variant (c.863C>T p.Ala288Val) in exon 2 of the telomerase reverse transcriptase (TERT) gene, a pathogenic variant in the IDH2 gene (c.419G>A p.Arg140Gln) in exon 4, and two uncertain significance variants (c.287A>G p.Tyr96Cys and c.413G>A p.Gly138Asp) in exons 3 and 4 of the Harvey rat sarcoma viral oncogene homolog (HRAS). P5 shows a nonsense variant in the neurofibromatosis 1 (NF1) gene (c.5242C>T p.Arg1748*) in exon 37. P6 harbors a pathogenic c.1799T>A p.Val600Glu in exon 15 of the BRAF gene. P9 shows a benign variant (c.532G>A p.Val178Ile) in the IDH1 gene. P10 presents an uncertain significance LP variant (c.1145G>A p.Arg382His) in exon 7 of the MSH2 gene. P11 carries a pathogenic variant (c.2341C>T p.Arg781*) in exon 9 of the ATRX gene and a pathogenic variant (c.1633G>A p.Glu545Lys) in exon 10 of the PIK3CA gene.
4. Discussion
In our study, we analyzed somatic variants in eleven pHGGs in a set of target genes involved in gliomagenesis [42,43,44]. The absence of genomic DNA did not allow us to establish the exclusive somatic origin of the variants to identify a potential cancer-predisposing syndrome.
Our results confirm the literature data, indicating TP53 and H3F3A as the most mutated genes in pHGGs (six patients each, 54%) [45,46] (Table 2).
The tumor suppressor and transcription factor p53 encoded by the T53 gene plays a critical role in pHGG tumorigenesis, and its deregulation promotes cell proliferation, migration, invasion, tumor cell stemness, and the failure of apoptosis in pHGG cells. In our study, TP53 variants were all associated with high-grade gliomas and other types of tumors (Table 2).
Variants in H3F3A are reported in a variety of solid tumors. We identified two recurrent pathogenic variants (Lys27Met (27%, P2, P5 and P11) and Gly34Arg (9%, P12)), a likely pathogenetic variant (Gly34Val) in one case (9%, P13), and an uncertain significance (VUS) LP variant (Asp82Glu) (9%; P3) (Table 2). c.83A>T (p.Lys27Met) is a pathogenic variant in exon 2 mainly associated with diffuse midline glioma (COSM327928). Gly34Arg and Gly34Val are pathogenic and likely-pathogenic variants associated with CNS brain tumors (COSM327929) and giant-cell tumor and astrocytoma (COSM502595), respectively. Moreover, Asp82Glu in exon 3 is reported as an uncertain significance LP, but is not present in the COSMIC database. While the role of the Lys27Met and Gly34Arg/Val variants in oncogenesis is sufficiently well-known, little is described regarding Asp82Glu. No data on frequency or specific tumors/clinical phenotype associations are reported. Interestingly Asp82 lies a region subject to post-translational modifications (two phosphorylation sites, Thr81 and Ser87, and a methylation/acetylation site, Lys80). It is not clearly known whether the substitution of aspartate with a glutamic acid, located between well-studied post-translational modification sites, could affect the final protein assembly or function. Future investigations will be indispensable in understanding the pathogenicity of Asp82Glu in the H3F3A gene. In two in three (67%) cases (P2 and P9) with Lys27Met we documented TP53 variants. TP53 variants and the consequent loss of functions are found in 70–80% of Lys27Met tumors, which contribute to radio resistance and poor prognosis [47,48].
The third most-often mutated gene in our series is EGFR (three cases, 27%) (Table 2). P4 shows only one pathogenic variant consisting of the insertion of nine nucleotides in exon 20 (c.2300_2301insCAGCGTGGA p.Ala767_Ser768insAlaTrpThr) in the essential C-helix domain (amino acids Ala767 to Cys775) [49]. This variant is not reported in databases but, in the same nucleotide position, different insertions are described that are associated with lung tumors [50,51]. P7 harbors VUS (c.1994G>A p.Gly665Asp and c.1973T>A p.Leu658Gln) which are exclusive to gliomas (COSM8256230 and COSM6354678, respectively). P11 harbor the VUS c.2024G>A p.Arg675Gln, which is reported in multiple tumors such as melanoma, and lung and prostate cancer, but not in brain tumors (COSM6938266). Meanwhile, it is reported in the HGMD database as a “Disease-causing mutation?” associated with central nervous system tumors (CM215217) [52]. The impossibility of studying the patient’s genomic DNA does not allow us to establish the germinal origin of the identified variant.
P3 shows a likely pathogenic Val658Ala in exon 14 and a benign Gly79Asp in exon 3 of PDGFRA, and P6 show a VUS Gly429Arg in exon 9 of PDGFRA. None of these variants is reported in the HGMD. Only the benign one, Gly79Asp, has been identified in several tumors, such as prostate tumors, myelodysplastic syndrome and meningioma (COSM5019287). This variant is inserted in a complex genetic pattern of variants (9 variants) of P3, and we cannot exclude that the combinations among the variants may contribute to causing a genetic background predisposing patients to gliomagenesis.
The tumor suppressor gene PTEN inhibits the PI3K/AKT pathway by suppressing cellular proliferation and survival. Inactivation of the PTEN gene can occur in high-grade gliomas. In our study, two patients (18%) show the genetic involvement of the PTEN gene with complete inactivation, through the combination of a single-nucleotide variant and the LOH of the wild type allele. In particular, P10 shows a pathogenic Iso101fs variant in exon 5 not reported in COSMIC, but associated with Cowden disease (CD110165), and P11 presents an unreported VUS Gln261Glu in exon 7 (Table 2).
Variants in the EZHIP, MSH2, IDH1, IDH2, (Telomerase reverse transcriptase) TERT, HRAS, NF1, BRAF, ATRX and PIK3CA genes are each identified in only one patient.
P1 shows a novel VUS (c.1328C>T p.Ser443Phe) in the serine-rich region of the C-terminus of the EZHIP gene. EZHIP is a single-exon gene with an unknown function, expressed mainly in the nucleus. EZHIP variants occur in posterior fossa group A (PFA) ependymomas (9.4%), endometrial cancers (5%) and melanoma (2.6%) [53]. Recently, Hübner J-M et al. identified that the C-terminus of EZHIP is critical in the interaction with PRC2 and its inhibition [54]. Future investigations will be indispensable in understanding the role of Ser443Phe.
P10 harbors a VUS LP variant (c.1145G>A p.Arg382His) in exon 7 of MSH2. This variant is associated only with lung adenocarcinoma (COSM3185920) and such as Disease-causing mutation in colorectal cancer (CM080440) [55]. In addition, P10 exhibits biallelic inactivation of both TP53 and PTEN genes, and also in this case a "sum" effect of all variants cannot be excluded.
P9 shows a benign variant (c.532G>A p.Val178Ile) in exon 6 of the IDH1 gene, with a frequency of 5% and 194 homozygotes (ExAC_nontcga_NFE) described in association with hematopoietic neoplasms, gliomas, thyroid tumors, and meningioma (COSM97131). To strengthen the neutral effect of c.532G>A, Ward et al. reported that Val178Ile retained the wild-type ability for isocitrate-dependent NADPH production without elevating cellular 2HG levels in the cell [56]; moreover, Thirumal Kumar et al. established the neutral impact of missense Val178Ile in the IDH1 homodimer region using in silico approaches [57].
P3 presents a complex pattern of variants, and we identified a new VUS (c.863C>T p.Ala288Val) in exon 2 of the TERT (Telomerase reverse transcriptase) gene, as well as a pathogenic variant in exon 4 of the IDH2 gene (c.419G>A p.Arg140Gln) associated with hematopoietic neoplasms/MDS, lung, stomach and upper aero-digestive tract tumors (COSM41590); however, these were never reported in brain tumors. Arg140 as Arg132 in the IDH1 gene and Arg172 in IDH2 lies in the active site of the IDH1/2 enzyme, suggesting a direct role of the variant on the catalytic niche [58]. Furthermore, P3 has two uncertain significance variants (c.287A>G p.Tyr96Cys and c.413G>A p.Gly138Asp) in exon 3 and 4 of the HRAS gene, not reported in COSMIC.
P5 shows a pathogenic nonsense variant, c.5242C>T p.Arg1748*, in exon 37, which is associated with NF1 (CM941094). The absence of P5 genomic DNA and specific clinical signs of NF1 does not exclude the presence of autosomal dominant NF1.
P6 has a pathogenic substitution, c.1799T>A p.Val600Glu, in exon 15 of the BRAF gene (COSM476). This variant has been mutated in many tumors such as melanoma, colorectal carcinoma, papillary carcinoma of thyroid and brain tumors, and Langerhans cell histiocytosis [59].
P11 has pathogenic variants in exon 9 of the ATRX (c.2341C>T p.Arg781*) and in exon 10 of the PIK3CA (c.1633G>A p.Glu545Lys) genes. c.2341C>T in ATRX is reported only in the COSMIC database, and is associated with melanoma and glioma (COSM1716656) instead. c.1633G>A in PIK3CA is reported to be associated with different tumors, including glioma (COSM763) and in megalencephaly-capillary malformation (CM126692).
For the molecular characterization of pHGGs, many integrated different diagnostic tools are required. In our study, the results derived from the gene panel were decisive for defining the molecular sub-type in two cases (P3 and P4), which were diagnosed as an uncertain sub type based on the routine diagnostic procedures (Table 1).
P3 (Figure 1) showed a pathogenic Arg140Gln variant in the IDH2 gene which, to the best of our knowledge, has never been reported in gliomas. Raynaud et al. searched for Arg140Gln on a series of 106 gliomas, and did not find this variant in any of the gliomas studied [60]. When the IDH2 variant occurs in gliomas, in almost all of the cases, it consists of a substitution of the arginine at codon 172, which is the exact residue analogous to Arg132 in IDH1. IDH2 variants are predominantly found in the oligodendrogliomas ATRX and TP53 wild type, TERT promoter mutated, and 1p–19q codeleted [19]. Our case is of special interest because, for the first time, Arg140Gln is documented in a glioma and occurred in a lesion with hybrid molecular features between astrocytoma and oligodendroglioma (with an absence of the ATRX variant, presence TP53 variants, and no 1p–19q codeletion) (Figure 1). It remains to be ascertained whether Arg140Gln in gliomas is just an anecdotal observation, whether it is typical in children, and whether it is associated with the same histotypes and molecular features as the most common IDH2 variants; lastly, its eventual diagnostic and prognostic significanceremains to be determined.
For patient P4 (Figure 2) the thalamic location and the age of the patient initially suggested a diagnosis of H3F3A-mutated gliomas. However, immunohistochemistry failed to stain for H3.3K27M-mutant protein, and successively, the gene panel analysis did not show any H3F3A variants. Consequently, this hypothesis was ruled out. Intriguingly, the immunostaining result for H3.3K27me3 was negative (Figure 2). FISH excluded MYCN, EGFR and PDGFR amplifications and CDKN2A deletion, while immunohistochemistry, followed by a gene panel, ruled out PXA-like pHGGs (no BRAF variants) (Table 2). A gene panel analysis, instead, documented an EGFR variant. Therefore, we diagnosed this case as a diffuse midline glioma EGFR mutant. This pHGG sub type is characterized by EZHIP epigenetic overexpression that, in turn, determines the loss of H3.3K27me3. Consequently, immunohistochemical demonstration of H3.3K27me3 loss is an effective method to indirectly ascertain its typical EZHIP overexpression [61].
Patients P1, P7 and P8 remained unclassified according to our integrated approach (Table 1).
Many causes can lead to a failure in the molecular classification of pHGGs, including the non-availability of all the necessary diagnostic tools (i.e., whole-genome methylation profiling) and the non-optimal quality of the tissues. In addition, some tumors could not fall within the currently described and classified molecular sub-types. The availability of molecular analyses reduces the number of unclassified cases.
5. Conclusions
TP53 and H3F3A were the most mutated genes. A novel variant in EZHIP and an IDH2 variant never reported in gliomas were documented.
The gene panel was crucial in recognizing the molecular features, determining the diagnosis in two cases (an IDH2 pathogenic variant in P3, and an EGFR variant in P4).
More than one-third of patients carry a variant in a gene associated with constitutional tumor-predisposing syndromes (PTEN, MSH2 and NF1).
Author Contributions
A.M.B. and L.G. (Laura Giunti) had equal contributions; L.G. (Laura Giunti), S.M., A.P. and F.C. conducted the experiment, prepared samples, and conducted downstream data analysis; A.M.B. and L.G. (Laura Giunti) prepared the drafts of the manuscript with feedback from M.S. and L.G. (Lorenzo Genitori) (neurosurgeons) A.P. (genetist), I.S. (neuro-oncologist) and C.C. (pathologist). All authors reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study. They approved the use of photographic figures and tumoral specimens for any subsequent genetic studies. The format has been authorized by our Institutional Review Board (code M0001).
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ostrom, Q.T.; de Blank, P.M.; Kruchko, C.; Petersen, C.M.; Liao, P.; Finlay, J.L.; Stearns, D.S.; Wolff, J.E.; Wolinsky, Y.; Letterio, J.J.; et al. Alex’s Lemonade Stand Foundation Infant and Childhood Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011. Neuro Oncol. 2015, 16 (Suppl. 10), x1–x36. [Google Scholar] [CrossRef] [PubMed]
- Sturm, D.; Pfister, S.M.; Jones, D.T.W. Pediatric Gliomas: Current Concepts on Diagnosis, Biology, and Clinical Management. J. Clin. Oncol. 2017, 35, 2370–2377. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro Oncol. 2021, 23 (Suppl. 3), iii1–iii105. [Google Scholar] [CrossRef] [PubMed]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537.e5. [Google Scholar] [CrossRef] [Green Version]
- Thorbinson, C.; Kilday, J.P. Childhood Malignant Brain Tumors: Balancing the Bench and Bedside. Cancers 2021, 13, 6099. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Torre, M.; Vasudevaraja, V.; Serrano, J.; DeLorenzo, M.; Malinowski, S.; Blandin, A.F.; Pages, M.; Ligon, A.H.; Dong, F.; Meredith, D.M.; et al. Molecular and clinicopathologic features of gliomas harboring NTRK fusions. Acta Neuropathol. Commun. 2020, 8, 107. [Google Scholar] [CrossRef]
- Mondal, G.; Lee, J.C.; Ravindranathan, A.; Villanueva-Meyer, J.E.; Tran, Q.T.; Allen, S.J.; Barreto, J.; Gupta, R.; Doo, P.; Van Ziffle, J.; et al. Pediatric bithalamic gliomas have a distinct epigenetic signature and frequent EGFR exon 20 insertions resulting in potential sensitivity to targeted kinase inhibition. Acta Neuropathol. 2020, 139, 1071–1088. [Google Scholar] [CrossRef]
- Kim, Y.Z. Altered histone modifications in gliomas. Brain Tumor Res. Treat. 2014, 2, 7–21. [Google Scholar] [CrossRef] [Green Version]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [Green Version]
- Castel, D.; Philippe, C.; Calmon, R.; Le Dret, L.; Truffaux, N.; Boddaert, N.; Pagès, M.; Taylor, K.R.; Saulnier, P.; Lacroix, L.; et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 2015, 130, 815–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orillac, C.; Thomas, C.; Dastagirzada, Y.; Hidalgo, E.T.; Golfinos, J.G.; Zagzag, D.; Wisoff, J.H.; Karajannis, M.A.; Snuderl, M. Pilocytic astrocytoma and glioneuronal tumor with histone H3 K27M mutation. Acta Neuropathol. Commun. 2016, 4, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, G.; Oberheim Bush, N.A.; Berger, M.S.; Perry, A.; Solomon, D.A. Diffuse non-midline glioma with H3F3A K27M mutation: A prognostic and treatment dilemma. Acta Neuropathol. Commun. 2017, 5, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Toenjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Bjerke, L.; Mackay, A.; Nandhabalan, M.; Burford, A.; Jury, A.; Popov, S.; Bax, D.; Carvalho, D.; Taylor, K.R.; Vinci, M.; et al. Histone H3.3. mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov. 2013, 3, 512–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, K.Y.; Won, J.K.; Park, C.K.; Kim, S.K.; Choi, S.H.; Kim, T.; Yun, H.; Park, S.H. H3 G34-mutant high-grade glioma. Brain Tumor Pathol. 2021, 38, 4–13. [Google Scholar] [CrossRef]
- Louis, D.N.; Wesseling, P.; Aldape, K.; Brat, D.J.; Capper, D.; Cree, I.A.; Eberhart, C.; Figarella-Branger, D.; Fouladi, M.; Fuller, G.N.; et al. cIMPACT-NOW update 6: New entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 2020, 30, 844–856. [Google Scholar] [CrossRef]
- Huang, J.; Yu, J.; Tu, L.; Huang, N.; Li, H.; Luo, Y. Isocitrate Dehydrogenase Mutations in Glioma: From Basic Discovery to Therapeutics Development. Front. Oncol. 2019, 9, 506. [Google Scholar] [CrossRef] [Green Version]
- Buccoliero, A.M.; Castiglione, F.; Degl’Innocenti, D.R.; Gheri, C.F.; Genitori, L.; Taddei, G.L. IDH1 mutation in pediatric gliomas: Has it a diagnostic and prognostic value? Fetal. Pediatr. Pathol. 2012, 31, 278–282. [Google Scholar] [CrossRef]
- Hartmann, C.; Meyer, J.; Balss, J.; Capper, D.; Mueller, W.; Christians, A.; Felsberg, J.; Wolter, M.; Mawrin, C.; Wick, W.; et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: A study of 1010 diffuse gliomas. Acta Neuropathol. 2009, 118, 469–474. [Google Scholar] [CrossRef] [Green Version]
- Sumerauer, D.; Krskova, L.; Vicha, A.; Misove, A.; Mamatjan, Y.; Jencova, P.; Vlckova, M.; Slamova, L.; Vanova, K.; Liby, P.; et al. Rare IDH1 variants are common in pediatric hemispheric diffuse astrocytomas and frequently associated with Li-Fraumeni syndrome. Acta Neuropathol. 2020, 139, 795–797. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, A.; Schrimpf, D.; Ryzhova, M.; Sturm, D.; Chavez, L.; Hovestadt, V.; Sharma, T.; Habel, A.; Burford, A.; Jones, C.; et al. H3-/IDH-wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathol. 2017, 134, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, A.; Ryzhova, M.; Hovestadt, V.; Bender, S.; Sturm, D.; Capper, D.; Meyer, J.; Schrimpf, D.; Kool, M.; Northcott, P.A.; et al. Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol. 2015, 129, 669–678. [Google Scholar] [CrossRef]
- Sturm, D.; Orr, B.A.; Toprak, U.H.; Hovestadt, V.; Jones, D.T.W.; Capper, D.; Sill, M.; Buchhalter, I.; Northcott, P.A.; Leis, I.; et al. New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 2016, 164, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Tauziède-Espariat, A.; Debily, M.A.; Castel, D.; Grill, J.; Puget, S.; Sabel, M.; Blomgren, K.; Gareton, A.; Dangouloff-Ros, V.; Lechapt, E.; et al. An integrative radiological, histopathological and molecular analysis of pediatric pontine histone-wildtype glioma with MYCN amplification (HGG-MYCN). Acta Neuropathol. Commun. 2019, 7, 87. [Google Scholar] [CrossRef]
- Koschmann, C.; Zamler, D.; MacKay, A.; Robinson, D.; Wu, Y.M.; Doherty, R.; Marini, B.; Tran, D.; Garton, H.; Muraszko, K.; et al. Characterizing and targeting PDGFRA alterations in pediatric high-grade glioma. Oncotarget 2016, 4, 65696–65706. [Google Scholar] [CrossRef] [Green Version]
- Paugh, B.S.; Qu, C.; Jones, C.; Liu, Z.; Adamowicz-Brice, M.; Zhang, J.; Bax, D.A.; Coyle, B.; Barrow, J.; Hargrave, D.; et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J. Clin. Oncol. 2010, 28, 3061–3068. [Google Scholar] [CrossRef]
- Li, J.; Liang, R.; Song, C.; Xiang, Y.; Liu, Y. Prognostic significance of epidermal growth factor receptor expression in glioma patients. Onco Targets Ther. 2018, 11, 731–742. [Google Scholar] [CrossRef] [Green Version]
- Viana-Pereira, M.; Lee, A.; Popov, S.; Bax, D.A.; Al-Sarraj, S.; Bridges, L.R.; Stávale, J.N.; Hargrave, D.; Jones, C.; Reis, R.M. Microsatellite instability in pediatric high grade glioma is associated with genomic profile and differential target gene inactivation. PLoS ONE 2011, 6, e20588. [Google Scholar] [CrossRef] [Green Version]
- Alphones, S.; Chatterjee, U.; Singh, A.; Das, A.; Zameer, L.; Achari, R.; Bhattacharya, A.; Roy, P. Immunohistochemical screening for mismatch repair protein deficiency in paediatric high-grade gliomas-institutional experience and review of literature. Childs Nerv. Syst. 2021, 37, 2521–2530. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.A.; Stechishin, O.D.; Luchman, H.A.; Lun, X.Q.; Senger, D.L.; Robbins, S.M.; Cairncross, J.G.; Weiss, S. Novel MSH6 mutations in treatment-naïve glioblastoma and anaplastic oligodendroglioma contribute to temozolomide resistance independently of MGMT promoter methylation. Clin. Cancer Res. 2014, 20, 4894–4903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quanye, S.; Chunying, P.; Qiuyuan, L.; Tianxiu, D.; Yucui, D.; Wenjing, X.; Peng, Z.; Yujiao, G.; Ziqi, Z.; Yifan, G.; et al. Up-regulation of MSH6 is associated with temozolomide resistance in human glioblastoma. Biochem. Biophys. Res. Commun. 2018, 496, 1040–1046. [Google Scholar]
- Yamashiro, K.; Nakao, K.; Ohba, S.; Hirose, Y. Human Glioma Cells Acquire Temozolomide Resistance After Repeated Drug Exposure Via DNA Mismatch Repair Dysfunction. Anticancer Res. 2020, 40, 1315–1323. [Google Scholar] [CrossRef]
- Sievers, P.; Sill, M.; Schrimpf, D.; Stichel, D.; Reuss, D.E.; Sturm, D.; Hench, J.; Frank, S.; Krskova, L.; Vicha, A.; et al. A subset of pediatric-type thalamic gliomas share a distinct DNA methylation profile, H3K27me3 loss and frequent alteration of EGFR. Neuro Oncol. 2021, 23, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Castel, D.; Kergrohen, T.; Tauziède-Espariat, A.; Mackay, A.; Ghermaoui, S.; Lechapt, E.; Pfister, S.M.; Kramm, C.M.; Boddaert, N.; Blauwblomme, T.; et al. Histone H3 wild-type DIPG/DMG overexpressing EZHIP extend the spectrum diffuse midline gliomas with PRC2 inhibition beyond H3-K27M mutation. Acta Neuropathol. 2020, 39, 1109–1113. [Google Scholar] [CrossRef]
- Jain, S.U.; Rashoff, A.Q.; Krabbenhoft, S.D.; Hoelper, D.; Do, T.J.; Gibson, T.J.; Lundgren, S.M.; Bondra, E.R.; Deshmukh, S.; Harutyunyan, A.S.; et al. H3 K27M and EZHIP Impede H3K27-Methylation Spreading by Inhibiting Allosterically Stimulated PRC2. Mol. Cell 2020, 80, 726–735.e7. [Google Scholar] [CrossRef]
- WHO Classification of Tumours Editorial Board. Central Nervous System Tumours. Lyon (France): International Agency for Research on Cancer, 5th ed.; WHO Classification of Tumours Series; WHO: Geneva, Switzerland, 2021; Volume 6. [Google Scholar]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point variants in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef] [Green Version]
- Feldman, A.Z.; Jennings, L.J.; Wadhwani, N.R.; Brat, D.J.; Horbinski, C.M. The Essentials of Molecular Testing in CNS Tumors: What to Order and How to Integrate Results. Curr. Neurol. Neurosci. Rep. 2020, 20, 23. [Google Scholar] [CrossRef]
- Braunstein, S.; Raleigh, D.; Bindra, R.; Mueller, S.; Haas-Kogan, D. Pediatric high-grade glioma: Current molecular landscape and therapeutic approaches. J. Neurooncol. 2017, 134, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Bready, D.; Placantonakis, D.G. Molecular Pathogenesis of Low-Grade Glioma. Neurosurg. Clin. N. Am. 2019, 30, 17–25. [Google Scholar] [CrossRef]
- Suri, V.; Das, P.; Pathak, P.; Jain, A.; Sharma, M.C.; Borkar, S.A.; Suri, A.; Gupta, D.; Sarkar, C. Pediatric glioblastomas: A histopathological and molecular genetic study. Neuro Oncol. 2009, 11, 274–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallappagoudar, S.; Yadav, R.K.; Lowe, B.R.; Partridge, J.F. Histone H3 mutations—A special role for H3.3 in tumorigenesis? Chromosoma 2015, 124, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, K.; Hoeman, C.; Becher, O. Children are not just little adults: Recent advances in understanding of diffuse intrinsic pontine glioma biology. Pediatr. Res. 2014, 75, 205–209. [Google Scholar] [CrossRef]
- Werbrouck, C.; Evangelista, C.C.S.; Lobón-Iglesias, M.J.; Barret, E.; Le Teuff, G.; Merlevede, J.; Brusini, R.; Kergrohen, T.; Mondini, M.; Bolle, S.; et al. TP53 Pathway Alterations Drive Radioresistance in Diffuse Intrinsic Pontine Gliomas (DIPG). Clin. Cancer Res. 2019, 25, 6788–6800. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, H.; Kobayashi, S.; Costa, D.B. EGFR exon 20 insertion mutations in non-small-cell lung cancer: Preclinical data and clinical implications. Lancet Oncol. 2012, 13, e23–e31. [Google Scholar] [CrossRef]
- Naidoo, J.; Sima, C.S.; Rodriguez, K.; Busby, N.; Nafa, K.; Ladanyi, M.; Riely, G.J.; Kris, M.G.; Arcila, M.E.; Yu, H.A. Epidermal growth factor receptor exon 20 insertions in advanced lung adenocarcinomas: Clinical outcomes and response to erlotinib. Cancer 2015, 121, 3212–3220. [Google Scholar] [CrossRef] [Green Version]
- Arcila, M.E.; Nafa, K.; Chaft, J.E.; Rekhtman, N.; Lau, C.; Reva, B.A.; Zakowski, M.F.; Kris, M.G.; Ladanyi, M. EGFR exon 20 insertion mutations in lung adenocarcinomas: Prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol. Cancer Ther. 2013, 12, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Akhavanfard, S.; Yehia, L.; Padmanabhan, R.; Reynolds, J.P.; Ni, Y.; Eng, C. Germline EGFR variants are over-represented in adolescents and young adults (AYA) with adrenocortical carcinoma. Hum. Mol. Genet. 2021, 29, 3679–3690. [Google Scholar] [CrossRef]
- Pajtler, K.W.; Wen, J.; Sill, M.; Lin, T.; Orisme, W.; Tang, B.; Hübner, J.M.; Ramaswamy, V.; Jia, S.; Dalton, J.D.; et al. Ellison Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. 2018, 13, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Hübner, J.M.; Müller, T.; Papageorgiou, D.N.; Mauermann, M.; Krijgsveld, J.; Russell, R.B.; Ellison, D.W.; Pfister, S.M.; Pajtler, K.W.; Kool, M. EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro Oncol. 2019, 21, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.Y.; Liu, X.; Li, V.K.; Ding, Y.; Yang, B.; Geng, J.; Lai, R.; Ding, S.; Ni, M.; Zhao, R. Detection of mismatch repair gene germline mutation carrier among Chinese population with colorectal cancer. BMC Cancer 2008, 8, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, P.S.; Cross, J.R.; Lu, C.; Weigert, O.; Abel-Wahab, O.; Levine, R.L.; Weinstock, D.M.; Sharp, K.A.; Thompson, C.B. Identification of additional IDH mutations associated with oncometabolite R(-)-2-hydroxyglutarate production. Oncogene 2012, 31, 2491–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thirumal Kumar, D.; Sneha, P.; Uppin, J.; Usha, S.; George Priya Doss, C. Investigating the Influence of Hotspot Mutations in Protein-Protein Interaction of IDH1 Homodimer Protein: A Computational Approach. Adv. Protein Chem. Struct. Biol. 2018, 111, 243–261. [Google Scholar]
- Yang, H.; Ye, D.; Guan, K.L.; Xiong, Y. IDH1 and IDH2 mutations in tumorigenesis: Mechanistic insights and clinical perspectives. Clin. Cancer Res. 2012, 18, 5562–5571. [Google Scholar] [CrossRef] [Green Version]
- Loo, E.; Khalili, P.; Beuhler, K.; Siddiqi, I.; Vasef, M.A. BRAF V600E Mutation Across Multiple Tumor Types: Correlation Between DNA-based Sequencing and Mutation-specific Immunohistochemistry. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 709–713. [Google Scholar] [CrossRef]
- Raynaud, S.; Carbuccia, N.; Colin, C.; Adélaïde, J.; Mozziconacci, M.J.; Metellus, P.; Chinot, O.; Birnbaum, D.; Chaffanet, M.; Figarella-Branger, D. Absence of R140Q mutation of isocitrate dehydrogenase 2 in gliomas and breast cancers. Oncol. Lett. 2010, 1, 883–884. [Google Scholar] [CrossRef]
- Nambirajan, A.; Sharma, A.; Rajeshwari, M.; Boorgula, M.T.; Doddamani, R.; Garg, A.; Suri, V.; Sarkar, C.; Sharma, M.C. EZH2 inhibitory protein (EZHIP/Cxorf67) expression correlates strongly with H3K27me3 loss in posterior fossa ependymomas and is mutually exclusive with H3K27M mutations. Brain Tumor Pathol. 2021, 38, 30–40. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).