
InsuLock: A Weakly Supervised Learning Approach for Accurate Insulator Prediction, and Variant Impact Quantification

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Preparation
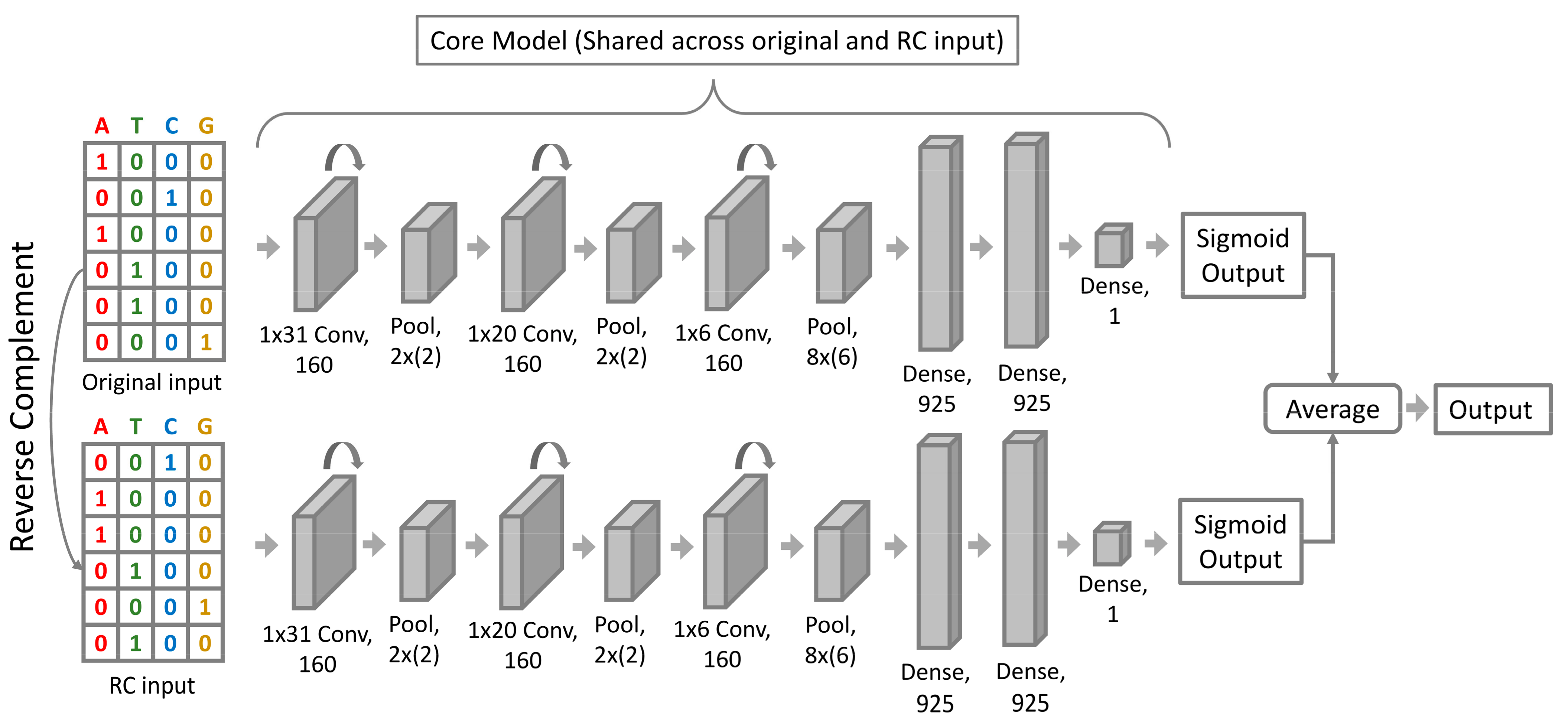
2.2. InsuLock Neural Network Architecture
2.3. Model Training and Evaluation
2.4. Weakly Supervised Object Detection via Gradient-Weighted Class Activation Mapping
2.5. Transcription Factor Motif Enrichment Analyses
2.6. Phylogenetic Conservation Analyses
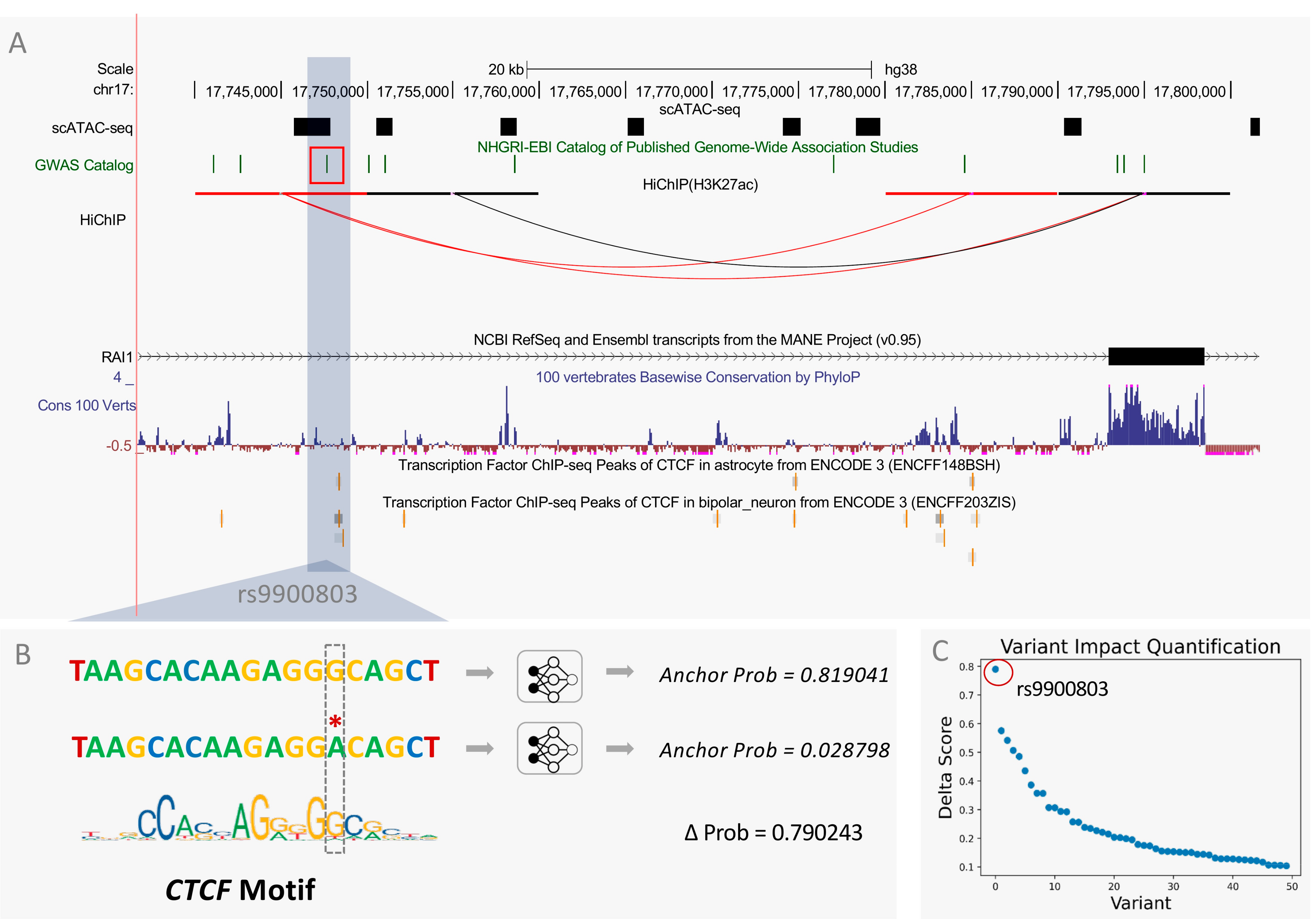
2.7. Variant Impact Quantification at Single-Cell Resolution
3. Results
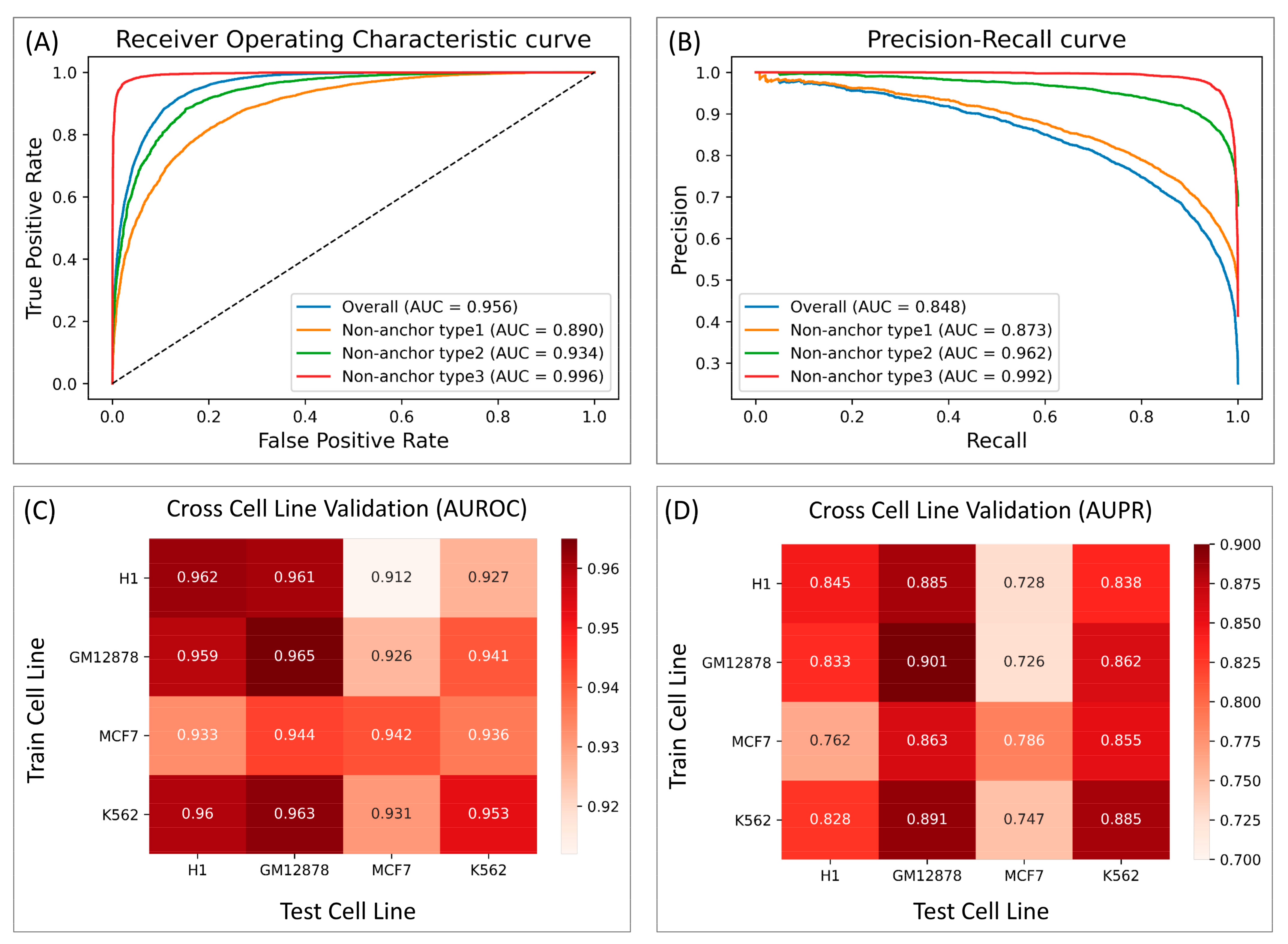
3.1. InsuLock Provides Accurate Insulator Predictions
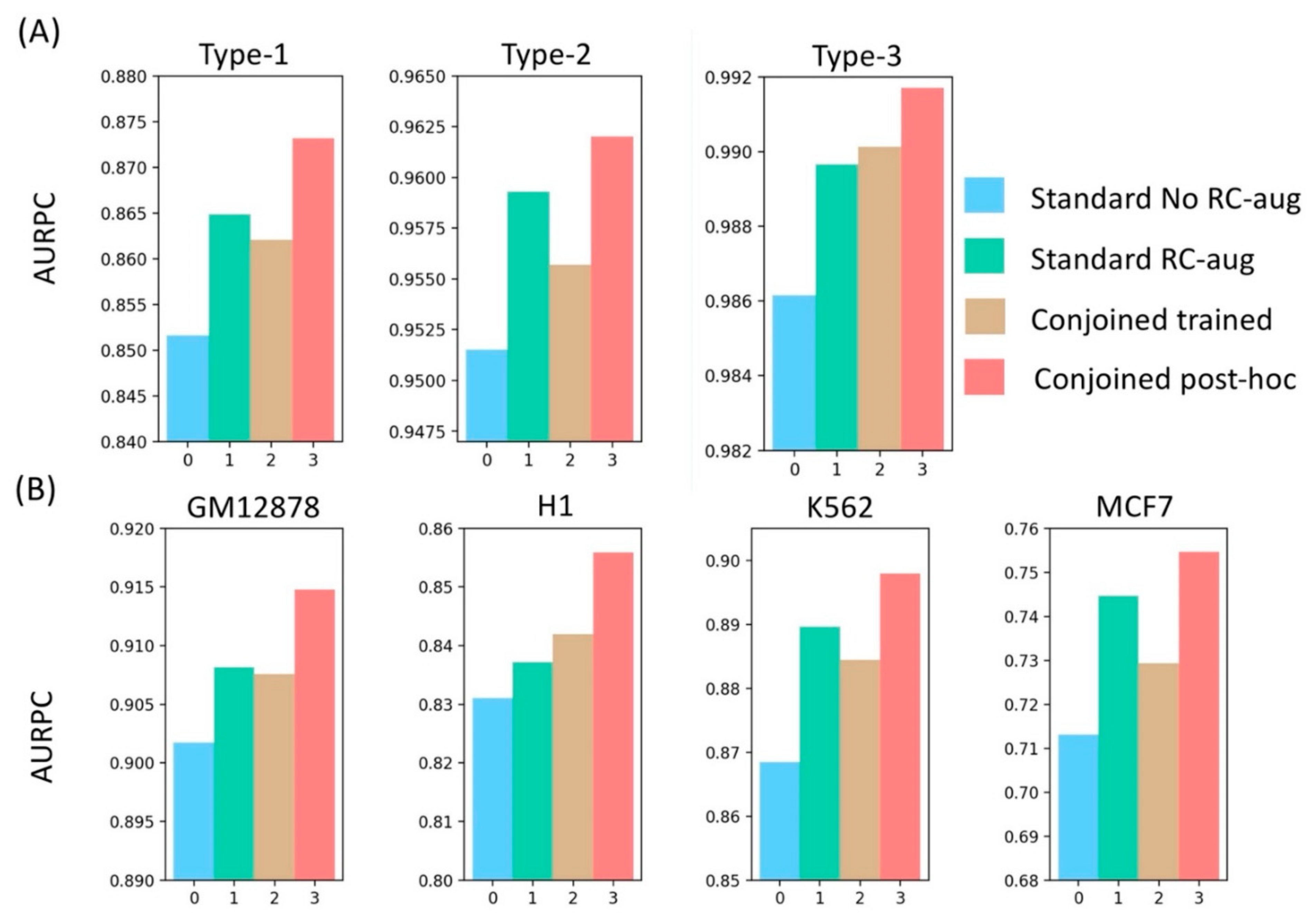
3.2. InsuLock’s Post-Hoc Siamese Model Outperform Standard Models
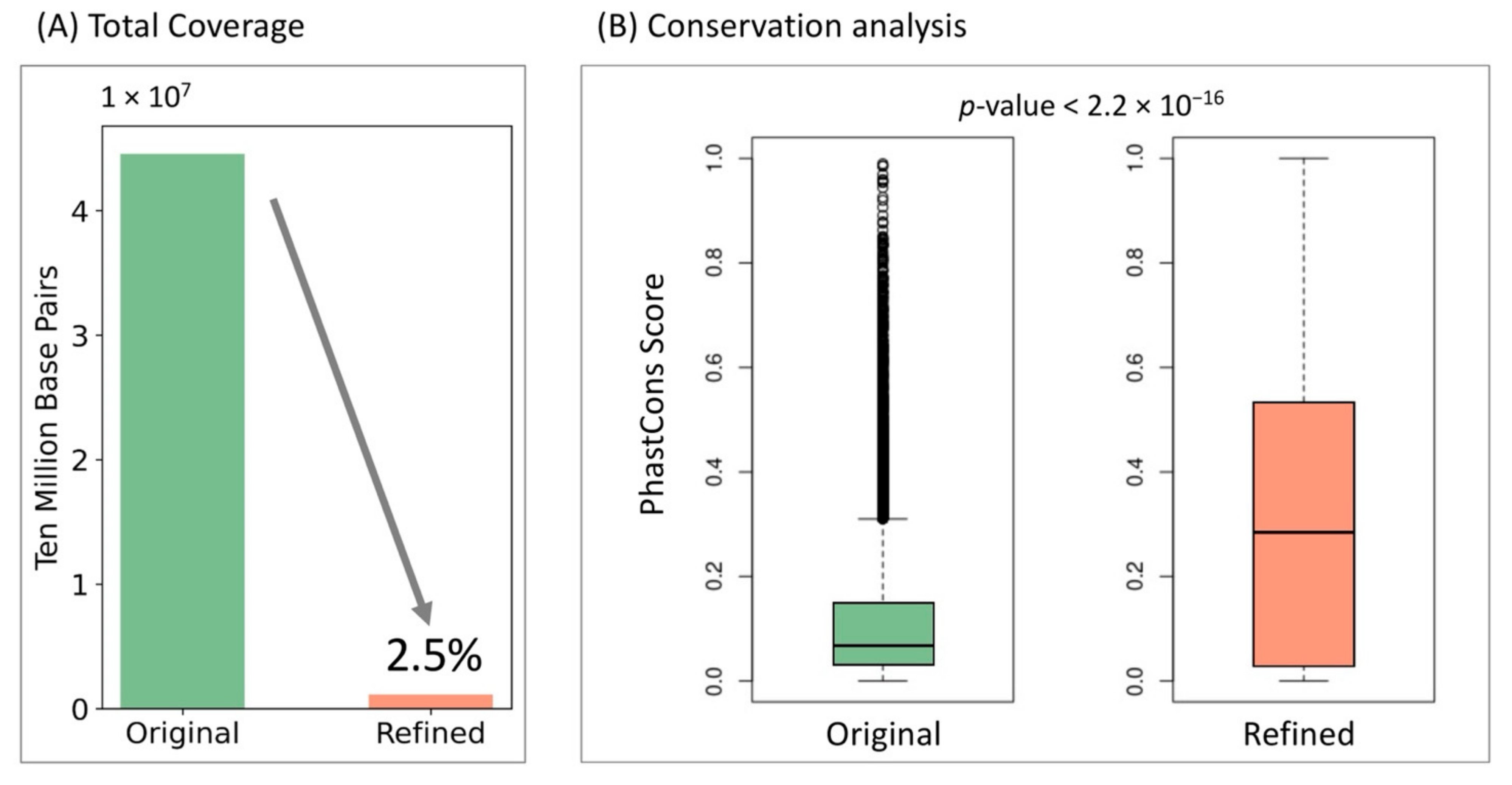
3.3. InsuLock’s Object Detection Module Accurately Refines Insulator Boundaries That Are Highly Conserved across Species
3.4. InsuLock Facilitates Precise Motif Discoveries to Uncover Novel TF Involvement to form Insulators
3.5. InsuLock Predicts Anchor Disruption Due to Functional Disease-Associated SNPs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kadauke, S.; Blobel, G.A. Chromatin loops in gene regulation. Biochim. Biophys. Acta 2009, 1789, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razin, S.V.; Ulianov, S.V. Gene functioning and storage within a folded genome. Cell. Mol. Biol. Lett. 2017, 22, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Xie, W. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 2019, 20, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Fullwood, M.J.; Ruan, Y. ChIP-based methods for the identification of long-range chromatin interactions. J. Cell. Biochem. 2009, 107, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Davidson, I.F.; Bauer, B.; Goetz, D.; Tang, W.; Wutz, G.; Peters, J.M. DNA loop extrusion by human cohesin. Science 2019, 366, 1338–1345. [Google Scholar] [CrossRef] [PubMed]
- Davidson, I.F.; Peters, J.M. Genome folding through loop extrusion by SMC complexes. Nat. Rev. Mol. Cell Biol. 2021, 22, 445–464. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suva, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef] [Green Version]
- Franke, M.; Ibrahim, D.M.; Andrey, G.; Schwarzer, W.; Heinrich, V.; Schopflin, R.; Kraft, K.; Kempfer, R.; Jerkovic, I.; Chan, W.L.; et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 2016, 538, 265–269. [Google Scholar] [CrossRef]
- Groschel, S.; Sanders, M.A.; Hoogenboezem, R.; de Wit, E.; Bouwman, B.A.M.; Erpelinck, C.; van der Velden, V.H.J.; Havermans, M.; Avellino, R.; van Lom, K.; et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 2014, 157, 369–381. [Google Scholar] [CrossRef] [Green Version]
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.L.; Bak, R.O.; Li, C.H.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A.; et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 2016, 351, 1454–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibn-Salem, J.; Kohler, S.; Love, M.I.; Chung, H.R.; Huang, N.; Hurles, M.E.; Haendel, M.; Washington, N.L.; Smedley, D.; Mungall, C.J.; et al. Deletions of chromosomal regulatory boundaries are associated with congenital disease. Genome Biol. 2014, 15, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, V.B.; Semple, C.A. When TADs go bad: Chromatin structure and nuclear organisation in human disease. F1000Research 2017, 6, PMC5373421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katainen, R.; Dave, K.; Pitkanen, E.; Palin, K.; Kivioja, T.; Valimaki, N.; Gylfe, A.E.; Ristolainen, H.; Hanninen, U.A.; Cajuso, T.; et al. CTCF/cohesin-binding sites are frequently mutated in cancer. Nat. Genet. 2015, 47, 818–821. [Google Scholar] [CrossRef]
- Kemp, C.J.; Moore, J.M.; Moser, R.; Bernard, B.; Teater, M.; Smith, L.E.; Rabaia, N.A.; Gurley, K.E.; Guinney, J.; Busch, S.E.; et al. CTCF haploinsufficiency destabilizes DNA methylation and predisposes to cancer. Cell Rep. 2014, 7, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Lettice, L.A.; Daniels, S.; Sweeney, E.; Venkataraman, S.; Devenney, P.S.; Gautier, P.; Morrison, H.; Fantes, J.; Hill, R.E.; FitzPatrick, D.R. Enhancer-adoption as a mechanism of human developmental disease. Hum. Mutat. 2011, 32, 1492–1499. [Google Scholar] [CrossRef]
- Lupianez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef] [Green Version]
- Northcott, P.A.; Lee, C.; Zichner, T.; Stutz, A.M.; Erkek, S.; Kawauchi, D.; Shih, D.J.; Hovestadt, V.; Zapatka, M.; Sturm, D.; et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 2014, 511, 428–434. [Google Scholar] [CrossRef]
- Weischenfeldt, J.; Dubash, T.; Drainas, A.P.; Mardin, B.R.; Chen, Y.; Stutz, A.M.; Waszak, S.M.; Bosco, G.; Halvorsen, A.R.; Raeder, B.; et al. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat. Genet. 2017, 49, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Kai, Y.; Andricovich, J.; Zeng, Z.; Zhu, J.; Tzatsos, A.; Peng, W. Predicting CTCF-mediated chromatin interactions by integrating genomic and epigenomic features. Nat. Commun. 2018, 9, 4221. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Wang, Y.; Yang, Y.; Zhang, Y.; Ma, J. Predicting CTCF-mediated chromatin loops using CTCF-MP. Bioinformatics 2018, 34, i133–i141. [Google Scholar] [CrossRef] [PubMed]
- Trieu, T.; Martinez-Fundichely, A.; Khurana, E. DeepMILO: A deep learning approach to predict the impact of non-coding sequence variants on 3D chromatin structure. Genome Biol. 2020, 21, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, H.; Dao, F.Y.; Zulfiqar, H.; Su, W.; Ding, H.; Liu, L.; Lin, H. A sequence-based deep learning approach to predict CTCF-mediated chromatin loop. Brief. Bioinform. 2021, 22, bbab031. [Google Scholar] [CrossRef]
- Kuang, S.; Wang, L. Deep Learning of Sequence Patterns for CCCTC-Binding Factor-Mediated Chromatin Loop Formation. J. Comput. Biol. 2021, 28, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.D.; Zhang, X.; Desai, K.; Aid, M.; Corradin, O.; Cowper-Sal Lari, R.; Akhtar-Zaidi, B.; Scacheri, P.C.; Haibe-Kains, B.; Lupien, M. ZNF143 provides sequence specificity to secure chromatin interactions at gene promoters. Nat. Commun. 2015, 2, 6186. [Google Scholar] [CrossRef]
- Beagan, J.A.; Duong, M.T.; Titus, K.R.; Zhou, L.; Cao, Z.; Ma, J.; Lachanski, C.V.; Gillis, D.R.; Phillips-Cremins, J.E. YY1 and CTCF orchestrate a 3D chromatin looping switch during early neural lineage commitment. Genome Res. 2017, 27, 1139–1152. [Google Scholar] [CrossRef] [Green Version]
- Arthur, R.K.; An, N.; Khan, S.; McNerney, M.E. The haploinsufficient tumor suppressor, CUX1, acts as an analog transcriptional regulator that controls target genes through distal enhancers that loop to target promoters. Nucleic Acids Res. 2017, 45, 6350–6361. [Google Scholar] [CrossRef]
- Zhang, J.; Lee, D.; Dhiman, V.; Jiang, P.; Xu, J.; McGillivray, P.; Yang, H.; Liu, J.; Meyerson, W.; Clarke, D.; et al. An integrative ENCODE resource for cancer genomics. Nat. Commun. 2020, 11, 3696. [Google Scholar] [CrossRef]
- Consortium, E.P.; Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Epstein, C.B.; Shoresh, N.; Adrian, J.; Kawli, T.; Davis, C.A.; Dobin, A.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [Green Version]
- Castro-Mondragon, J.A.; Riudavets-Puig, R.; Rauluseviciute, I.; Berhanu Lemma, R.; Turchi, L.; Blanc-Mathieu, R.; Lucas, J.; Boddie, P.; Khan, A.; Manosalva Perez, N.; et al. JASPAR 2022: The 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2022, 50, D165–D173. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L. Pytorch: An imperative style, high-performance deep learning library. Adv. Neural Inf. Process. Syst. 2019, 32, 8026–8037. [Google Scholar]
- Selvaraju, R.R.; Cogswell, M.; Das, A.; Vedantam, R.; Parikh, D.; Batra, D. Grad-cam: Visual explanations from deep networks via gradient-based localization. In Proceedings of the IEEE International Conference on Computer Vision, Venice, Italy, 22–29 October 2017; pp. 618–626. [Google Scholar]
- Bailey, T.L.; Machanick, P. Inferring direct DNA binding from ChIP-seq. Nucleic Acids Res. 2012, 40, e128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [Green Version]
- Corces, M.R.; Shcherbina, A.; Kundu, S.; Gloudemans, M.J.; Fresard, L.; Granja, J.M.; Louie, B.H.; Eulalio, T.; Shams, S.; Bagdatli, S.T.; et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat. Genet. 2020, 52, 1158–1168. [Google Scholar] [CrossRef]
- Cao, Z.; Zhang, S. Simple tricks of convolutional neural network architectures improve DNA-protein binding prediction. Bioinformatics 2019, 35, 1837–1843. [Google Scholar] [CrossRef]
- Granja, J.M.; Corces, M.R.; Pierce, S.E.; Bagdatli, S.T.; Choudhry, H.; Chang, H.Y.; Greenleaf, W.J. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 2021, 53, 403–411. [Google Scholar] [CrossRef]
- Fang, R.; Preissl, S.; Li, Y.; Hou, X.; Lucero, J.; Wang, X.; Motamedi, A.; Shiau, A.K.; Zhou, X.; Xie, F.; et al. Comprehensive analysis of single cell ATAC-seq data with SnapATAC. Nat. Commun. 2021, 12, 1337. [Google Scholar] [CrossRef]
- Justice, M.; Carico, Z.M.; Stefan, H.C.; Dowen, J.M. A WIZ/Cohesin/CTCF Complex Anchors DNA Loops to Define Gene Expression and Cell Identity. Cell Rep. 2020, 31, 107503. [Google Scholar] [CrossRef] [PubMed]
- Whalen, S.; Truty, R.M.; Pollard, K.S. Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat. Genet. 2016, 48, 488–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubert, F.; Srivas, R.; Spacek, D.V.; Kasowski, M.; Ruiz-Velasco, M.; Sinnott-Armstrong, N.; Greenside, P.; Narasimha, A.; Liu, Q.; Geller, B.; et al. Landscape of cohesin-mediated chromatin loops in the human genome. Nature 2020, 583, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Tam, V.; Patel, N.; Turcotte, M.; Bosse, Y.; Pare, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Slager, R.E.; Newton, T.L.; Vlangos, C.N.; Finucane, B.; Elsea, S.H. Mutations in RAI1 associated with Smith-Magenis syndrome. Nat. Genet. 2003, 33, 466–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girirajan, S.; Elsas, L.J., 2nd; Devriendt, K.; Elsea, S.H. RAI1 variations in Smith-Magenis syndrome patients without 17p11.2 deletions. J. Med. Genet. 2005, 42, 820–828. [Google Scholar] [CrossRef] [Green Version]
- Carmona-Mora, P.; Walz, K. Retinoic Acid Induced 1, RAI1: A Dosage Sensitive Gene Related to Neurobehavioral Alterations Including Autistic Behavior. Curr. Genom. 2010, 11, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Haybaeck, J.; Postruznik, M.; Miller, C.L.; Dulay, J.R.; Llenos, I.C.; Weis, S. Increased expression of retinoic acid-induced gene 1 in the dorsolateral prefrontal cortex in schizophrenia, bipolar disorder, and major depression. Neuropsychiatr. Dis. Treat. 2015, 11, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Jing, F.; Zhang, S.W.; Cao, Z.; Zhang, S. An Integrative Framework for Combining Sequence and Epigenomic Data to Predict Transcription Factor Binding Sites Using Deep Learning. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 18, 355–364. [Google Scholar] [CrossRef]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srinivasan, S.S.; Gong, Y.; Xu, S.; Hwang, A.; Xu, M.; Girgenti, M.J.; Zhang, J. InsuLock: A Weakly Supervised Learning Approach for Accurate Insulator Prediction, and Variant Impact Quantification. Genes 2022, 13, 621. https://doi.org/10.3390/genes13040621
Srinivasan SS, Gong Y, Xu S, Hwang A, Xu M, Girgenti MJ, Zhang J. InsuLock: A Weakly Supervised Learning Approach for Accurate Insulator Prediction, and Variant Impact Quantification. Genes. 2022; 13(4):621. https://doi.org/10.3390/genes13040621
Chicago/Turabian StyleSrinivasan, Shushrruth Sai, Yanwen Gong, Siwei Xu, Ahyeon Hwang, Min Xu, Matthew J. Girgenti, and Jing Zhang. 2022. "InsuLock: A Weakly Supervised Learning Approach for Accurate Insulator Prediction, and Variant Impact Quantification" Genes 13, no. 4: 621. https://doi.org/10.3390/genes13040621
APA StyleSrinivasan, S. S., Gong, Y., Xu, S., Hwang, A., Xu, M., Girgenti, M. J., & Zhang, J. (2022). InsuLock: A Weakly Supervised Learning Approach for Accurate Insulator Prediction, and Variant Impact Quantification. Genes, 13(4), 621. https://doi.org/10.3390/genes13040621