
New Insights on Gene by Environmental Effects of Drugs of Abuse in Animal Models Using GeneNetwork


Abstract
1. Introduction
2. Materials and Methods
2.1. Phenotype Data
2.2. New Genotypes from Sequencing
2.3. Genome-Wide Efficient Mixed Model Association (GEMMA), Kinship within the BXD Strains, and QTL Mapping
2.4. Identification of Novel QTLs
2.5. QTL Confidence Intervals
2.6. Cis-eQTL Mapping
2.7. “Gene Friends”, or Co-Expression Analysis
2.8. Gene Variant Analysis
2.9. PheWAS
3. Results
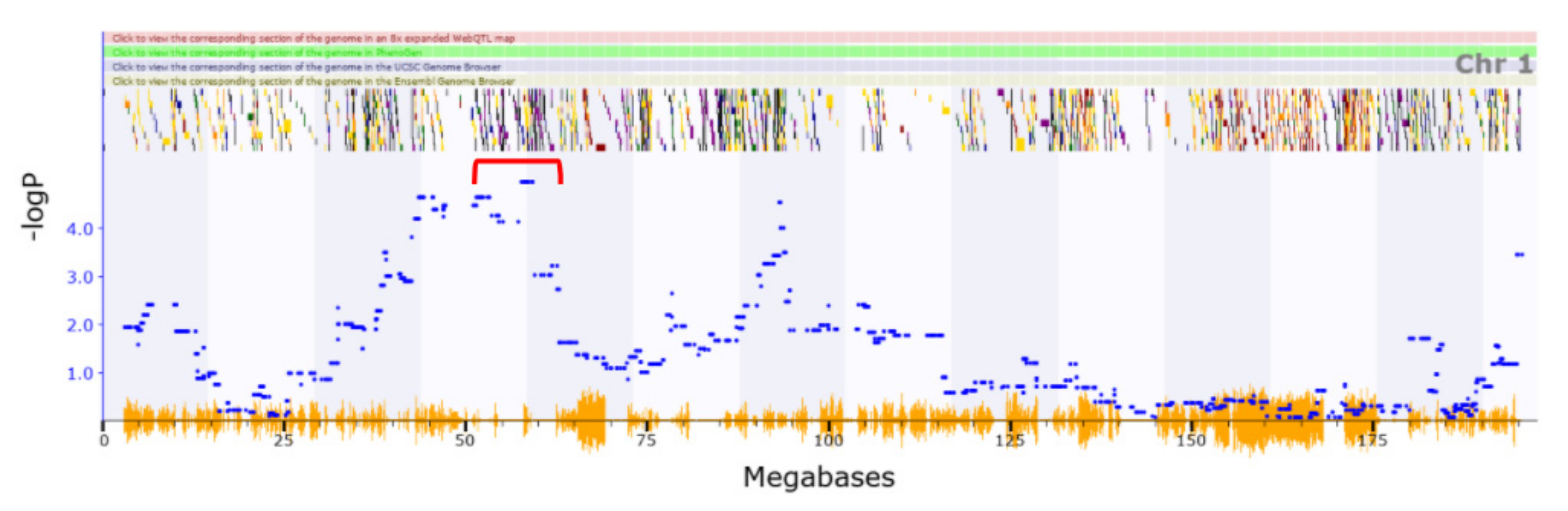
3.1. Identification of QTLs
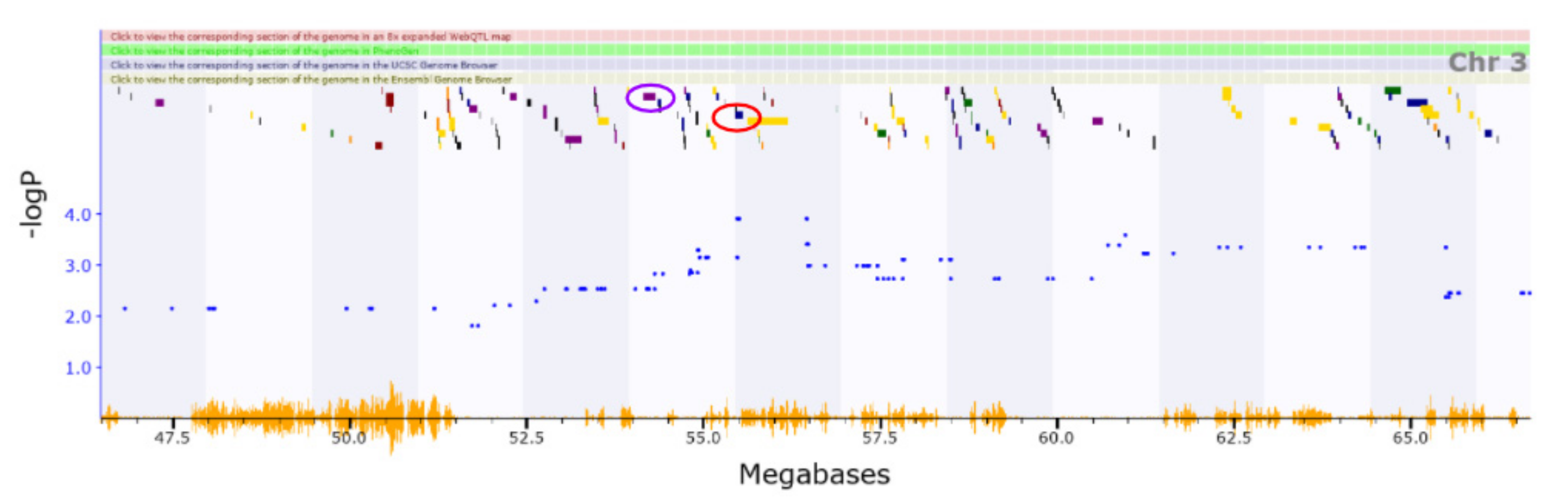
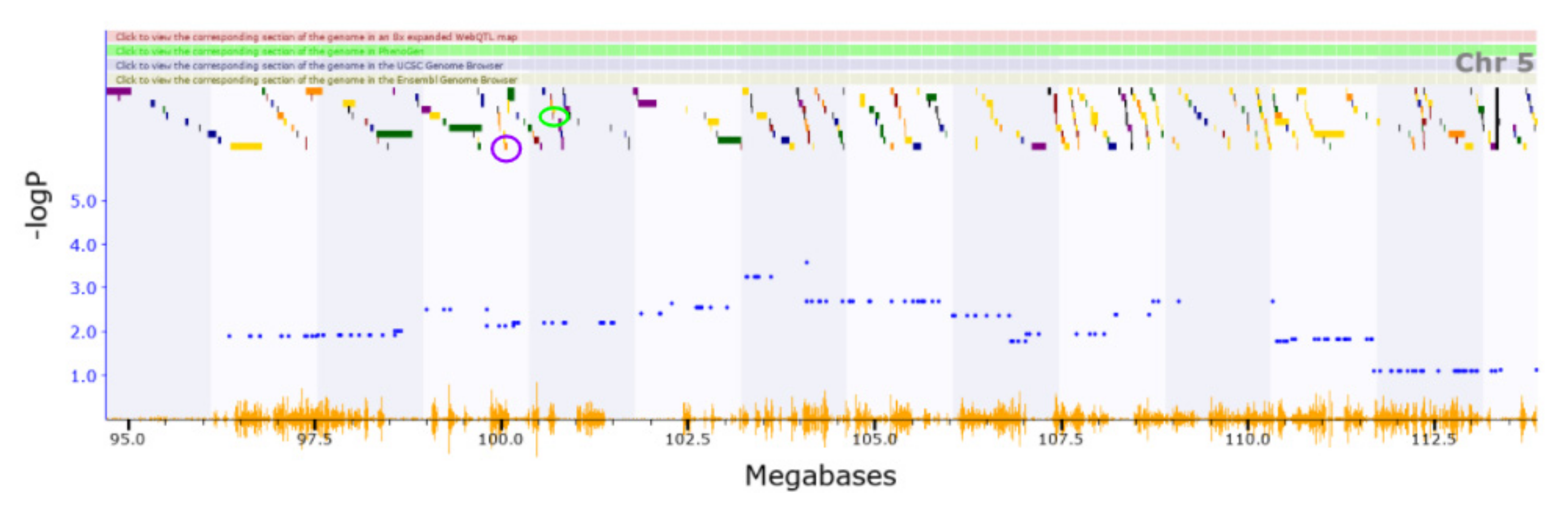
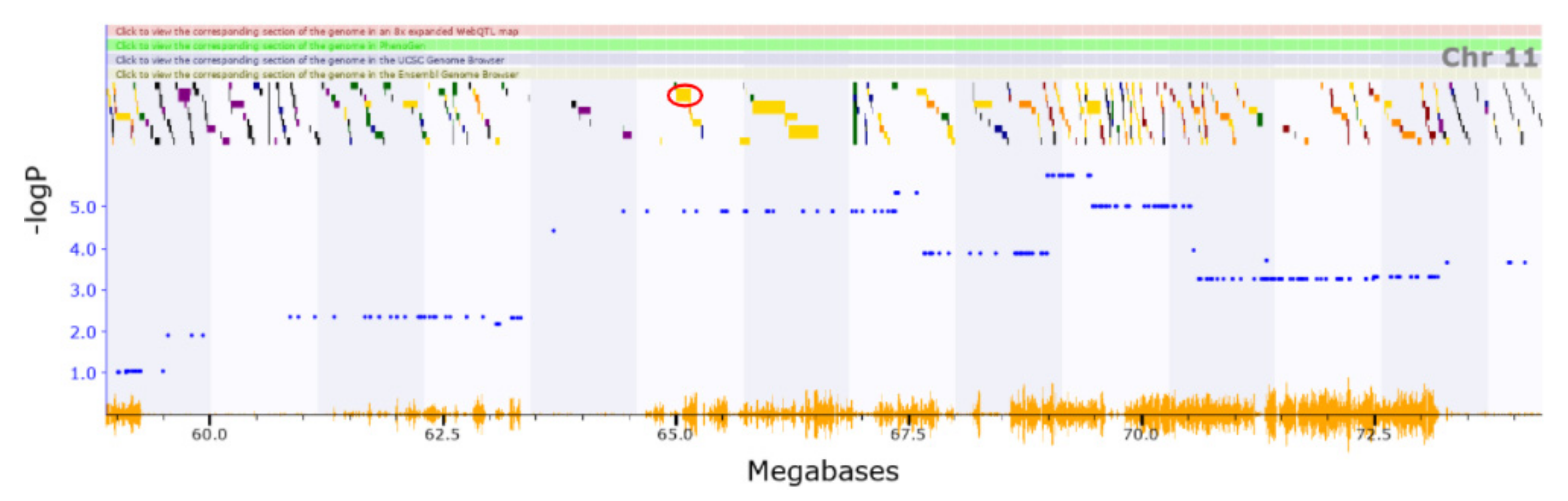
3.2. Novel QTL
3.3. Candidate Causal Genes within Novel QTL
3.4. Co-Expression Networks or “Gene-Friends”
3.5. Gene Variant Analysis
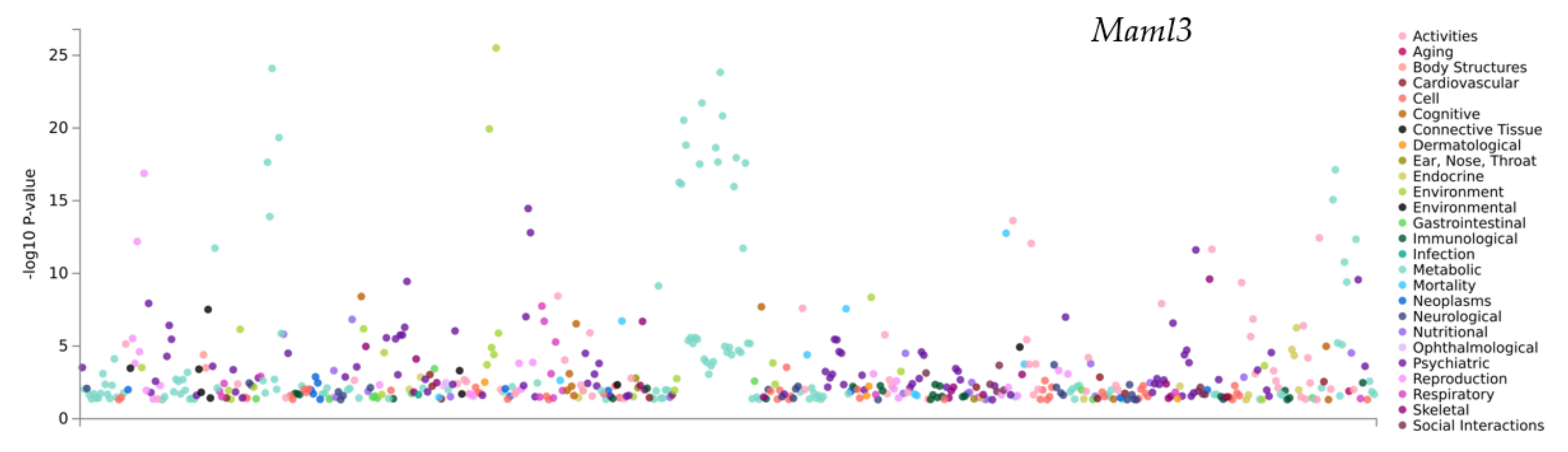
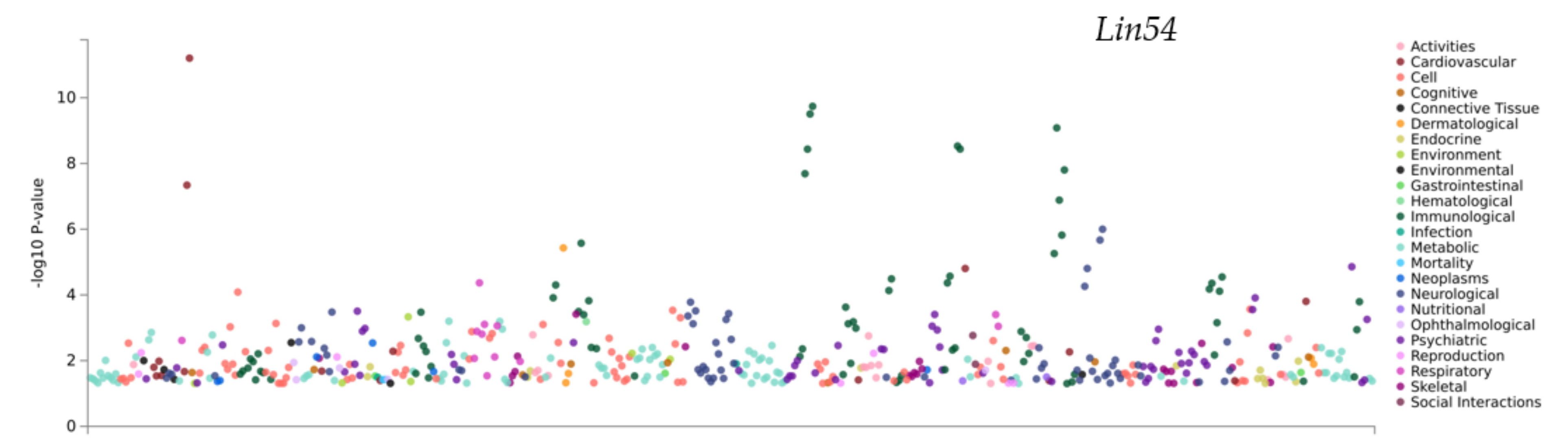
3.6. PheWAS Analysis of the Genes within QTLs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef]
- Ashbrook, D.G.; Arends, D.; Prins, P.; Mulligan, M.K.; Roy, S.; Williams, E.G.; Lutz, C.M.; Valenzuela, A.; Bohl, C.J.; Ingels, J.F.; et al. A platform for experimental precision medicine: The extended BXD mouse family. Cell Syst. 2021, 12, 235–247.e9. [Google Scholar] [CrossRef]
- Castle, W.E. Variation in the hooded pattern of rats, and a new allele of hooded. Genetics 1951, 36, 254–266. [Google Scholar] [CrossRef]
- Belknap, J.K.; Crabbe, J.C.; Plomin, R.; McClearn, G.E.; Sampson, K.E.; O’Toole, L.A.; Gora-Maslak, G. Single-locus control of saccharin intake in BXD/Ty recombinant inbred (RI) mice: Some methodological implications for RI strain analysis. Behav. Genet. 1992, 22, 81–100. [Google Scholar] [CrossRef]
- Belknap, J.K.; Metten, P.; Helms, M.L.; O’Toole, L.A.; Angeli-Gade, S.; Crabbe, J.C.; Phillips, T.J. Quantitative trait loci (QTL) applications to substances of abuse: Physical dependence studies with nitrous oxide and ethanol in BXD mice. Behav. Genet. 1993, 23, 213–222. [Google Scholar] [CrossRef]
- Dickson, P.E.; Miller, M.M.; Calton, M.A.; Bubier, J.A.; Cook, M.N.; Goldowitz, D.; Chesler, E.J.; Mittleman, G. Systems genetics of intravenous cocaine self-administration in the BXD recombinant inbred mouse panel. Psychopharmacology 2016, 233, 701–714. [Google Scholar] [CrossRef]
- Grisel, J.E.; Belknap, J.K.; O’Toole, L.A.; Helms, M.L.; Wenger, C.D.; Crabbe, J.C. Quantitative trait loci affecting methamphetamine responses in BXD recombinant inbred mouse strains. J. Neurosci. 1997, 17, 745–754. [Google Scholar] [CrossRef]
- Palmer, A.A.; Lessov-Schlaggar, C.N.; Ponder, C.A.; McKinnon, C.S.; Phillips, T.J. Sensitivity to the locomotor-stimulant effects of ethanol and allopregnanolone: A quantitative trait locus study of common genetic influence. Genes Brain Behav. 2006, 5, 506–517. [Google Scholar] [CrossRef]
- Phillips, T.J.; Belknap, J.K.; Buck, K.J.; Cunningham, C.L. Genes on mouse chromosomes 2 and 9 determine variation in ethanol consumption. Mamm. Genome 1998, 9, 936–941. [Google Scholar]
- Rodriguez, L.A.; Plomin, R.; Blizard, D.A.; Jones, B.C.; McClearn, G.E. Alcohol acceptance, preference, and sensitivity in mice. I. Quantitative genetic analysis using BXD recombinant inbred strains. Alcohol. Clin. Exp. Res. 1994, 18, 1416–1422. [Google Scholar] [CrossRef]
- Boon, A.C.M.; Williams, R.W.; Sinasac, D.S.; Webby, R.J. A novel genetic locus linked to pro-inflammatory cytokines after virulent H5N1 virus infection in mice. BMC Genom. 2014, 15, 1017. [Google Scholar] [CrossRef]
- Grizzle, W.E.; Mountz, J.D.; Yang, P.-A.; Xu, X.; Sun, S.; Van Zant, G.E.; Williams, R.W.; Hsu, H.C.; Zhang, H.G. BXD recombinant inbred mice represent a novel T cell-mediated immune response tumor model. Int. J. Cancer 2002, 101, 270–279. [Google Scholar] [CrossRef]
- Hayes, K.S.; Hager, R.; Grencis, R.K. Sex-dependent genetic effects on immune responses to a parasitic nematode. BMC Genom. 2014, 15, 193. [Google Scholar] [CrossRef]
- Miyairi, I.; Ziebarth, J.; Laxton, J.D.; Wang, X.; van Rooijen, N.; Williams, R.W.; Lu, L.; Byrne, G.I.; Cui, Y. Host genetics and Chlamydia disease: Prediction and validation of disease severity mechanisms. PLoS ONE 2012, 7, e33781. [Google Scholar] [CrossRef]
- Wang, J.; Yoon, T.W.; Read, R.; Yi, A.-K.; Williams, R.W.; Fitzpatrick, E.A. Genetic variability of T cell responses in Hypersensitivity Pneumonitis identified using the BXD genetic reference panel. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L631–L643. [Google Scholar] [CrossRef]
- Ashbrook, D.G.; Sharmin, N.; Hager, R. Offspring genes indirectly influence sibling and maternal behavioral strategies over resource share. Proc. R. Soc. B Biol. Sci. 2017, 284, 20171059. [Google Scholar]
- Ashbrook, D.G.; Roy, S.; Clifford, B.G.; Riede, T.; Scattoni, M.L.; Heck, D.H.; Lu, L.; Williams, R.W. Born to cry: A genetic dissection of infant vocalization. Front. Behav. Neurosci. 2018, 12, 250. [Google Scholar] [CrossRef]
- Dickson, P.E.; Roy, T.A.; McNaughton, K.A.; Wilcox, T.D.; Kumar, P.; Chesler, E.J. Systems genetics of sensation seeking. Genes Brain Behav. 2019, 18, e12519. [Google Scholar] [CrossRef]
- Graybeal, C.; Bachu, M.; Mozhui, K.; Saksida, L.M.; Bussey, T.J.; Sagalyn, E.; Williams, R.W.; Holmes, A. Strains and stressors: An analysis of touchscreen learning in genetically diverse mouse strains. PLoS ONE 2014, 9, e87745. [Google Scholar] [CrossRef]
- Knoll, A.T.; Jiang, K.; Levitt, P. Quantitative trait locus mapping and analysis of heritable variation in affiliative social behavior and co-occurring traits. Genes Brain Behav. 2017, 17, e12431. [Google Scholar] [CrossRef]
- Li, Z.; Mulligan, M.K.; Wang, X.; Miles, M.F.; Lu, L.; Williams, R.W. A transposon in Comt generates mRNA variants and causes widespread expression and behavioral differences among mice. PLoS ONE 2010, 5, e12181. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, M.K.; Abreo, T.; Neuner, S.M.; Parks, C.; Watkins, C.E.; Houseal, M.T.; Shapaker, T.M.; Hook, M.; Tan, H.; Wang, X. Identification of a Functional Non-coding Variant in the GABA A Receptor α2 Subunit of the C57BL/6J Mouse Reference Genome: Major Implications for Neuroscience Research. Front. Genet. 2019, 10, 188. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.G.; Mouchiroud, L.; Frochaux, M.; Pandey, A.; Andreux, P.A.; Deplancke, B.; Auwerx, J. An evolutionarily conserved role for the aryl hydrocarbon receptor in the regulation of movement. PLoS Genet. 2014, 10, e1004673. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.; Williams, R.W.; Auwerx, J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013, 497, 451–457. [Google Scholar] [CrossRef]
- Neuner, S.M.; Garfinkel, B.P.; Wilmott, L.A.; Ignatowska-Jankowska, B.M.; Citri, A.; Orly, J.; Lu, L.; Overall, R.W.; Mulligan, M.K.; Kempermann, G. Systems genetics identifies Hp1bp3 as a novel modulator of cognitive aging. Neurobiol. Aging 2016, 46, 58–67. [Google Scholar] [CrossRef][Green Version]
- Roy, S.; Sleiman, M.B.; Jha, P.; Williams, E.G.; Ingels, J.F.; Chapman, C.J.; McCarty, M.S.; Ziebarth, J.D.; Hook, M.; Sun, A. Gene-by-environment modulation of lifespan and weight gain in the murine BXD family. Nat. Metab. 2021, 3, 1217–1227. [Google Scholar] [CrossRef]
- Sandoval-Sierra, J.V.; Helbing, A.H.B.; Williams, E.G.; Ashbrook, D.G.; Roy, S.; Williams, R.W.; Mozhui, K. Body weight and high-fat diet are associated with epigenetic aging in female members of the BXD murine family. Aging Cell. 2020, 19, e13207. [Google Scholar] [CrossRef]
- Williams, E.G.; Pfister, N.; Roy, S.; Statzer, C.; Ingels, J.; Bohl, C.; Hasan, M.; Čuklina, J.; Bühlmann, P.; Zamboni, N.; et al. Multiomic profiling of the liver across diets and age in a diverse mouse population. Cell Syst. 2022, 13, 43–57.e6. [Google Scholar] [CrossRef]
- Neuner, S.M.; Heuer, S.E.; Huentelman, M.J.; O’Connell, K.M.S.; Kaczorowski, C.C. Harnessing Genetic Complexity to Enhance Translatability of Alzheimer’s Disease Mouse Models: A Path toward Precision Medicine. Neuron 2019, 101, 399–411.e5. [Google Scholar] [CrossRef]
- Neuner, S.M.; Heuer, S.E.; Zhang, J.-G.; Philip, V.M.; Kaczorowski, C.C. Identification of Pre-symptomatic Gene Signatures That Predict Resilience to Cognitive Decline in the Genetically Diverse AD-BXD Model. Front. Genet. 2019, 10, 35. [Google Scholar] [CrossRef]
- Neuner, S.M.; Wilmott, L.A.; Hoffmann, B.R.; Mozhui, K.; Kaczorowski, C.C. Hippocampal proteomics defines pathways associated with memory decline and resilience in normal aging and Alzheimer’s disease mouse models. Behav. Brain Res. 2017, 322, 288–298. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, K.M.S.; Ouellette, A.R.; Neuner, S.M.; Dunn, A.R.; Kaczorowski, C.C. Genetic background modifies CNS-mediated sensorimotor decline in the AD-BXD mouse model of genetic diversity in Alzheimer’s disease. Genes Brain Behav. 2019, 18, e12603. [Google Scholar] [CrossRef] [PubMed]
- Rosen, G.D.; Pung, C.J.; Owens, C.B.; Caplow, J.; Kim, H.; Mozhui, K.; Lu, L.; Williams, R.W. Genetic modulation of striatal volume by loci on Chrs 6 and 17 in BXD recombinant inbred mice. Genes Brain Behav. 2009, 8, 296–308. [Google Scholar] [CrossRef] [PubMed]
- McKnite, A.M.; Perez-Munoz, M.E.; Lu, L.; Williams, E.G.; Brewer, S.; Andreux, P.A.; Bastiaansen, J.W.M.; Wang, X.; Kachman, S.D.; Auwerx, J. Murine gut microbiota is defined by host genetics and modulates variation of metabolic traits. PLoS ONE 2012, 7, e3919. [Google Scholar] [CrossRef]
- Taylor, B.A.; Heiniger, H.J.; Meier, H. Genetic analysis of resistance to cadmium-induced testicular damage in mice. Proceedings of the Society for Experimental Biology and Medicine. Soc. Exp. Biol. Med. 1973, 143, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.A.; Wnek, C.; Kotlus, B.S.; Roemer, N.; MacTaggart, T.; Phillips, S.J. Genotyping new BXD recombinant inbred mouse strains and comparison of BXD and consensus maps. Mamm. Genome 1999, 10, 335–348. [Google Scholar] [CrossRef]
- Peirce, J.L.; Lu, L.; Gu, J.; Silver, L.M.; Williams, R.W. A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC Genet. 2004, 5, 7. [Google Scholar] [CrossRef]
- Wang, J.; Williams, R.W.; Manly, K.F. WebQTL: Web-based complex trait analysis. Neuroinformatics 2003, 1, 299–308. [Google Scholar] [CrossRef]
- Philip, V.M.; Duvvuru, S.; Gomero, B.; Ansah, T.A.; Blaha, C.D.; Cook, M.N.; Hamre, K.M.; Lariviere, W.R.; Matthews, D.B.; Mittleman, G. High-throughput behavioral phenotyping in the expanded panel of BXD recombinant inbred strains. Genes Brain Behav. 2010, 9, 129–159. [Google Scholar] [CrossRef]
- Broman, K.W.; Gatti, D.M.; Simecek, P.; Furlotte, N.A.; Prins, P.; Sen, Ś.; Yandell, B.S.; Churchill, G.A. R/qtl2: Software for Mapping Quantitative Trait Loci with High-Dimensional Data and Multiparent Populations. Genetics 2019, 211, 495–502. [Google Scholar] [CrossRef]
- Manichaikul, A.; Dupuis, J.; Sen, S.; Broman, K.W. Poor Performance of Bootstrap Confidence Intervals for the Location of a Quantitative Trait Locus. Genetics 2006, 174, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Duncan, D.; Shi, Z.; Zhang, B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): Update 2013. Nucleic Acids Res. 2013, 41, W77–W83. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Vasaikar, S.; Shi, Z.; Greer, M.; Zhang, B. WebGestalt 2017: A more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017, 45, W130–W137. [Google Scholar] [CrossRef]
- Zhang, B.; Kirov, S.; Snoddy, J. WebGestalt: An integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005, 33, W741–W748. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Eddy, S.R. Where did the BLOSUM62 alignment score matrix come from? Nat. Biotechnol. 2004, 22, 1035–1036. [Google Scholar] [CrossRef]
- Watanabe, K.; Stringer, S.; Frei, O.; Umićević Mirkov, M.; de Leeuw, C.; Polderman, T.J.C.; van der Sluis, S.; Andreassen, O.A.; Neale, B.M.; Posthuma, D. A global overview of pleiotropy and genetic architecture in complex traits. Nat. Genet. 2019, 51, 1339–1348. [Google Scholar] [CrossRef]
- Haley, C.; Knott, S. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 1992, 69, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Eppig, J.T. Mouse genome informatics (MGI) resource: Genetic, genomic, and biological knowledgebase for the laboratory mouse. ILAR J. 2017, 58, 17–41. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.; Markel, P.D.; Beeson, M.A.; Gordon, L.G.; Johnson, T.E. Mapping quantitative trait loci for ethanol-induced anesthesia in LSxSS recombinant inbred and F2 mice: Methodology and results. Alcohol Alcohol. 1994, 2, 79–86. [Google Scholar]
- Le Roy, I.; Perez-Diaz, F.; Cherfouh, A.; Roubertoux, P.L. Preweanling sensorial and motor development in laboratory mice: Quantitative trait loci mapping. Dev. Psychobiol. 1999, 34, 139–158. [Google Scholar] [CrossRef]
- Nakamura, K.; Xiu, Y.; Ohtsuji, M.; Sugita, G.; Abe, M.; Ohtsuji, N.; Hamano, Y.; Jiang, Y.; Takahashi, N.; Shirai, T. Genetic dissection of anxiety in autoimmune disease. Hum. Mol. Genet. 2003, 12, 1079–1086. [Google Scholar] [CrossRef][Green Version]
- Håvik, B.; Degenhardt, F.A.; Johansson, S.; Fernandes, C.P.D.; Hinney, A.; Scherag, A.; Lybæk, H.; Djurovic, S.; Christoforou, A.; Ersland, K.M. DCLK1 variants are associated across schizophrenia and attention deficit/hyperactivity disorder. PLoS ONE 2012, 7, e35424. [Google Scholar] [CrossRef]
- Nair, H.K.; Hain, H.; Quock, R.M.; Philip, V.M.; Chesler, E.J.; Belknap, J.K.; Lariviere, W.R. Genomic loci and candidate genes underlying inflammatory nociception. Pain 2011, 152, 599–606. [Google Scholar] [CrossRef]
- Riccio, A.; Li, Y.; Tsvetkov, E.; Gapon, S.; Yao, G.L.; Smith, K.S.; Engin, E.; Rudolph, U.; Bolshakov, V.Y.; Clapham, D.E. Decreased anxiety-like behavior and Gαq/11-dependent responses in the amygdala of mice lacking TRPC4 channels. J. Neurosci. 2014, 34, 3653–3667. [Google Scholar] [CrossRef]
- Mardones, M.D.; Andaur, G.A.; Varas-Godoy, M.; Henriquez, J.F.; Salech, F.; Behrens, M.I.; Couve, A.; Inestrosa, N.C.; Varela-Nallar, L. Frizzled-1 receptor regulates adult hippocampal neurogenesis. Mol. Brain 2016, 9, 29. [Google Scholar] [CrossRef]
- Barth, A.; Bilkei-Gorzo, A.; Drews, E.; Otte, D.M.; Diaz-Lacava, A.; Varadarajulu, J.; Turck, C.W.; Wienker, T.F.; Zimmer, A. Analysis of quantitative trait loci in mice suggests a role of Enoph1 in stress reactivity. J. Neurochem. 2014, 128, 807–817. [Google Scholar] [CrossRef]
- Mikasa, M.; Kanai, K.; Li, Y.; Yoshino, H.; Mogushi, K.; Hayashida, A.; Ikeda, A.; Kawajiri, S.; Okuma, Y.; Kashihara, K. COQ2 variants in Parkinson’s disease and multiple system atrophy. J. Neural Transm. 2018, 125, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Funato, H.; Miyoshi, C.; Fujiyama, T.; Kanda, T.; Sato, M.; Wang, Z.; Ma, J.; Nakane, S.; Tomita, J.; Ikkyu, A. Forward-genetics analysis of sleep in randomly mutagenized mice. Nature 2016, 539, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, Y.; Fujita, E.; Yu, Z.; Yamagata, T.; Momoi, M.Y.; Momoi, T.; Onaka, T. Impairment of social and emotional behaviors in Cadm1-knockout mice. Biochem. Biophys. Res. Commun. 2010, 396, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Sarowar, T.; Grabrucker, S.; Föhr, K.; Mangus, K.; Eckert, M.; Bockmann, J.; Boeckers, T.M.; Grabrucker, A.M. Enlarged dendritic spines and pronounced neophobia in mice lacking the PSD protein RICH2. Mol. Brain 2016, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Katayama, K.; Okamoto, T.; Yamada, K.; Takashima, N.; Nagao, S.; Aruga, J. Impaired auditory-vestibular functions and behavioral abnormalities of Slitrk6-deficient mice. PLoS ONE 2011, 6, e16497. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, S.; Cordeiro, R.; Craig, T.; van Dam, J.; Wood, S.H.; de Magalhães, J.P. GeneFriends: An online co-expression analysis tool to identify novel gene targets for aging and complex diseases. BMC Genom. 2012, 13, 535. [Google Scholar] [CrossRef]
- Van Dam, S.; Craig, T.; de Magalhães, J.P. GeneFriends: A human RNA-seq-based gene and transcript co-expression database. Nucleic Acids Res. 2015, 43, D1124–D1132. [Google Scholar] [CrossRef]
- Wang, J.C.; Foroud, T.; Hinrichs, A.L.; Le, N.X.H.; Bertelsen, S.; Budde, J.P.; Harari, O.; Koller, D.L.; Wetherill, L.; Agrawal, A. A genome-wide association study of alcohol-dependence symptom counts in extended pedigrees identifies C15orf53. Mol. Psychiatry 2013, 18, 1218–1224. [Google Scholar] [CrossRef]
- Howard, D.M.; Adams, M.J.; Clarke, T.-K.; Hafferty, J.D.; Gibson, J.; Shirali, M.; Coleman, J.R.I.; Hagenaars, S.P.; Ward, J.; Wigmore, E.M. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 2019, 22, 343–352. [Google Scholar] [CrossRef]
- Okbay, A.; Baselmans, B.M.L.; De Neve, J.-E.; Turley, P.; Nivard, M.G.; Fontana, M.A.; Meddens, S.F.W.; Linnér, R.K.; Rietveld, C.A.; Derringer, J. Genetic variants associated with subjective well-being, depressive symptoms, and neuroticism identified through genome-wide analyses. Nat. Genet. 2016, 48, 624–633. [Google Scholar] [CrossRef]
- Ripke, S.; Wray, N.R.; Lewis, C.M.; Hamilton, S.P.; Weissman, M.M.; Breen, G.; Byrne, E.M.; Blackwood, D.H.; Boomsma, D.I.; Cichon, S. A mega-analysis of genome-wide association studies for major depressive disorder. Mol. Psychiatry 2013, 18, 497–511. [Google Scholar] [PubMed]
- Day, F.R.; Ong, K.K.; Perry, J.R.B. Elucidating the genetic basis of social interaction and isolation. Nat. Commun. 2018, 9, 2457. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.E.; Tyrrell, J.; Wood, A.R.; Beaumont, R.N.; Ruth, K.S.; Tuke, M.A.; Yaghootkar, H.; Hu, Y.; Teder-Laving, M.; Hayward, C. Genome-Wide Association Analyses in 128,266 Individuals Identifies New Morningness and Sleep Duration Loci. PLoS Genet. 2016, 12, e1006125. [Google Scholar] [CrossRef] [PubMed]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Bipolar Disorder and Schizophrenia Working Group of the Psychiatric Genomics Consortium & Bipolar Disorder and Schizophrenia Working Group of the Psychiatric Genomics Consortium. Genomic dissection of bipolar disorder and schizophrenia, including 28 subphenotypes. Cell 2018, 173, 1705–1715.e16. [Google Scholar] [CrossRef]
- Fung, H.-C.; Scholz, S.; Matarin, M.; Simón-Sánchez, J.; Hernandez, D.; Britton, A.; Gibbs, J.R.; Langefeld, C.; Stiegert, M.L.; Schymick, J. Genome-wide genotyping in Parkinson’s disease and neurologically normal controls: First stage analysis and public release of data. Lancet Neurol. 2006, 5, 911–916. [Google Scholar] [CrossRef]
- Proenca, C.C.; Gao, K.P.; Shmelkov, S.V.; Rafii, S.; Lee, F.S. Slitrks as emerging candidate genes involved in neuropsychiatric disorders. Trends Neurosci. 2011, 34, 143–153. [Google Scholar] [CrossRef]
- Beaubien, F.; Cloutier, J.-F. Differential expression of Slitrk family members in the mouse nervous system. Dev. Dyn. 2009, 238, 3285–3296. [Google Scholar] [CrossRef]
- Jin, J.; Liu, L.; Chen, W.; Gao, Q.; Li, H.; Wang, Y.; Qian, Q. The Implicated Roles of Cell Adhesion Molecule 1 (CADM1) Gene and Altered Prefrontal Neuronal Activity in Attention-Deficit/Hyperactivity Disorder: A “Gene-Brain-Behavior Relationship”? Front. Genet. 2019, 10, 882. [Google Scholar] [CrossRef]
- Bou Khalil, R.; Khoury, E.; Richa, S. The Comorbidity of Fibromyalgia Syndrome and Attention Deficit and Hyperactivity Disorder from a Pathogenic Perspective. Pain Med. 2018, 19, 1705–1709. [Google Scholar] [CrossRef]
- Treister, R.; Eisenberg, E.; Demeter, N.; Pud, D. Alterations in pain response are partially reversed by methylphenidate (Ritalin) in adults with attention deficit hyperactivity disorder (ADHD). Pain Pract. 2015, 15, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Stray, L.L.; Kristensen, Ø.; Lomeland, M.; Skorstad, M.; Stray, T.; Tønnessen, F.E. Motor regulation problems and pain in adults diagnosed with ADHD. Behav. Brain Funct. 2013, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Van Breda, S.G.J.; Claessen, S.M.H.; van Herwijnen, M.; Theunissen, D.H.J.; Jennen, D.G.J.; de Kok, T.M.C.M.; Kleinjans, J.C.S. Integrative omics data analyses of repeated dose toxicity of valproic acid in vitro reveal new mechanisms of steatosis induction. Toxicology 2018, 393, 160–170. [Google Scholar] [CrossRef]
- Schulpen, S.H.W.; Pennings, J.L.A.; Piersma, A.H. Gene expression regulation and pathway analysis after valproic acid and carbamazepine exposure in a human embryonic stem cell based neuro-developmental toxicity assay. Toxicol. Sci. 2015, 146, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Rempel, E.; Hoelting, L.; Waldmann, T.; Balmer, N.V.; Schildknecht, S.; Grinberg, M.; Das Gaspar, J.A.; Shinde, V.; Stöber, R.; Marchan, R. A transcriptome-based classifier to identify developmental toxicants by stem cell testing: Design, validation and optimization for histone deacetylase inhibitors. Arch. Toxicol. 2015, 89, 1599–1618. [Google Scholar] [CrossRef] [PubMed]
- Reuterswärd, P.; Bergström, S.; Orikiiriza, J.; Lindquist, E.; Bergström, S.; Andersson Svahn, H.; Ayoglu, B.; Uhlén, M.; Wahlgren, M.; Normark, J. Levels of human proteins in plasma associated with acute paediatric malaria. Malar. J. 2018, 17, 426. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome | QTL Confidence Interval (Mb) | Summary of Phenotype | Relevant Behaviour Phenotype | PMID of Relevant Phenotype |
|---|---|---|---|---|
| Chr1 | 37.671–78.94 | Locomotion | Loss of righting induced by ethanol | 8974320 |
| Chr1 | 37.671–78.94 | Locomotion | Vertical clinging | 10086232 |
| Chr1 | 68.798–80.329 | Cocaine and locomotion | Loss of righting induced by ethanol | 16803863 |
| Chr1 | 91.214–99.884 | Vertical activity | Loss of righting induced by ethanol | 16803863 |
| Chr3 | 51.723–56.473 | Vertical activity | ||
| Chr7 | 97.466–104.149 | |||
| Chr12 | 82.859–96.105 | BXD_11407 | ||
| Chr14 | 109.994–114.751 | BXD_12023 | ||
| Chr15 | 71.035–77.148 | Motor coordination, anxiety | Abnormal fear/anxiety-related behaviour | 10556431 |
| Number of Variants | Type of Variant | Gene |
|---|---|---|
| 2 | Missense | Ccdc169 |
| 1 | Missense | Ccna1 |
| 1 | In-frame insertion | Dclk1 |
| 2 | Frameshift | Frem2 |
| 1 | Stop loss | Frem2 |
| 9 | Missense | Frem2 |
| 1 | Frameshift | Mab21l1 |
| 6 | Missense | Mab21l1 |
| 9 | Frameshift | Nbea |
| 2 | In-frame deletions | Nbea |
| 19 | Missense | Nbea |
| 3 | Stop loss | Nbea |
| 4 | In-frame deletions | Postn |
| 1 | In-frame insertions | Postn |
| 6 | Missense | Postn |
| 1 | Start loss | Postn |
| 1 | Frameshift | Spg20 |
| 8 | Missense | Spg20 |
| 1 | Stop gain | Spg20 |
| 1 | Stop loss | Spg20 |
| 1 | Missense | Trpc4 |
| Number of Variants | Type of Variant | Gene |
|---|---|---|
| 3 | Frameshift | Cops4 |
| 3 | Missense | Cops4 |
| 1 | Missense | Enoph1 |
| 1 | Stop gain | Enoph1 |
| 1 | Frameshift | Hnrnpd |
| 5 | Missense | Hnrnpd |
| 3 | Missense | Hnrnpd |
| 1 | Splice donor | Hnrnpd |
| 2 | Frameshift | Hpse |
| 1 | In-frame insertion | Hpse |
| 13 | Missense | Hpse |
| 1 | Splice donor | Hpse |
| 3 | Missense | Lin54 |
| 1 | Frameshift | Sec31a |
| 1 | In-frame deletion | Sec31a |
| 1 | Splice donor | Sec31a |
| 1 | Missense | Tmem150c |
| 1 | Splice donor | Tmem150c |
| Number of Variants | Type of Variant | Gene |
|---|---|---|
| 1 | Frameshift | 4931429L15Ri |
| 2 | Missense | 4931429L15Ri |
| 1 | Stop gain | 4931429L15Ri |
| 2 | Frameshift | Cadm1 |
| 5 | Missense | Cadm1 |
| 1 | In-frame deletion | Cep164 |
| 3 | Missense | Cep164 |
| 1 | Stop loss | Cep164 |
| atlas ID | PMID | Year | Domain | Trait | p-Value | N |
|---|---|---|---|---|---|---|
| 4314 | 30643251 | 2019 | Psychiatric | Ever smoked regulary | 3.47 × 10−15 | 262990 |
| 3654 | 31427789 | 2019 | Psychiatric | Smoking status: Never | 2.45 × 10−12 | 384964 |
| 4327 | 30643256 | 2019 | Psychiatric | Well-being spectrum | 2.73 × 10−10 | 2311184 |
| 4322 | 30643256 | 2019 | Psychiatric | Depressive symptoms (univariate) | 3.58 × 10−10 | 1067913 |
| 4313 | 30643251 | 2019 | Psychiatric | Age of initiation of regular smoking | 1.16 × 10−8 | 632802 |
| 3425 | 31427789 | 2019 | Psychiatric | Ever smoked | 9.55 × 10−8 | 385013 |
| 3236 | 31427789 | 2019 | Psychiatric | Past tobacco smoking | 1.02 × 10−7 | 355594 |
| 4274 | 30846698 | 2019 | Psychiatric | Short sleep | 2.64 × 10−7 | 411934 |
| 3261 | 31427789 | 2019 | Psychiatric | Alcohol intake frequency | 3.80 × 10−7 | 386082 |
| 4326 | 30643256 | 2019 | Psychiatric | Depressive symptoms (MA GWAMA) | 5.12 × 10−7 | 1067913 |
| 56 | 27089181 | 2016 | Psychiatric | Depressive symptoms | 1.72 × 10−6 | 161460 |
| 3796 | 29942085 | 2018 | Psychiatric | Depressive symptoms | 1.75 × 10−6 | 381455 |
| 3235 | 31427789 | 2019 | Psychiatric | Current tobacco smoking | 2.71 × 10−6 | 386150 |
| 4293 | 30718901 | 2019 | Psychiatric | Depression | 3.19 × 10−6 | 500199 |
| 3268 | 31427789 | 2019 | Psychiatric | Alcohol intake versus 10 years previously | 3.41 × 10−6 | 357907 |
| 4171 | 29970889 | 2018 | Psychiatric | Loneliness | 3.44 × 10−6 | 445024 |
| 4170 | 29970889 | 2018 | Psychiatric | Loneliness (MTAG) | 3.72 × 10−6 | 487647 |
| atlas ID | PMID | Year | Domain | Trait | p-Value | N |
|---|---|---|---|---|---|---|
| 4327 | 30643256 | 2019 | Psychiatric | Well-being spectrum | 1.40 × 10−5 | 2311184 |
| 3998 | 29500382 | 2018 | Psychiatric | Tense | 1.23 × 10−5 | 263635 |
| 3291 | 31427789 | 2019 | Psychiatric | Tense | 2.80 × 10−4 | 374129 |
| 4293 | 30718901 | 2019 | Psychiatric | Depression | 3.12 × 10−4 | 500199 |
| 4325 | 30643256 | 2019 | Psychiatric | Neuroticism (MA GWAMA) | 3.94 × 10−4 | 523783 |
| 3798 | 29942085 | 2018 | Psychiatric | Worry subcluster | 5.57 × 10−4 | 348219 |
| 4087 | 29255261 | 2018 | Psychiatric | Neuroticism | 8.90 × 10−4 | 329821 |
| 4322 | 30643256 | 2019 | Psychiatric | Depressive symptoms (univariate) | 1.05 × 10−3 | 1067913 |
| 3301 | 31427789 | 2019 | Psychiatric | Seen doctor (GP) for nerves, anxiety, tension or depression | 1.11 × 10−3 | 383771 |
| 4321 | 30643256 | 2019 | Psychiatric | Neuroticism (univariate) | 1.12 × 10−3 | 523783 |
| 4326 | 30643256 | 2019 | Psychiatric | Depressive symptoms (MA GWAMA) | 1.27 × 10−3 | 1067913 |
| 3302 | 31427789 | 2019 | Psychiatric | Seen a psychiatrist for nerves, anxiety, tension or depression | 2.48 × 10−3 | 384700 |
| 3745 | 31427789 | 2019 | Psychiatric | Happiness and subjective well-being—General happiness | 2.83 × 10−3 | 126132 |
| 4011 | 29662059 | 2018 | Psychiatric | Broad depression | 3.34 × 10−3 | 322580 |
| 4013 | 29662059 | 2018 | Psychiatric | Major depressive disorder (ICD-coded) | 3.46 × 10−3 | 217584 |
| 4269 | 30867560 | 2019 | Psychiatric | Neuroticism general factor | 3.84 × 10−3 | 270059 |
| 3230 | 31427789 | 2019 | Psychiatric | Morning/evening person (chronotype) | 4.42 × 10−3 | 345148 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chunduri, A.; Watson, P.M.; Ashbrook, D.G. New Insights on Gene by Environmental Effects of Drugs of Abuse in Animal Models Using GeneNetwork. Genes 2022, 13, 614. https://doi.org/10.3390/genes13040614
Chunduri A, Watson PM, Ashbrook DG. New Insights on Gene by Environmental Effects of Drugs of Abuse in Animal Models Using GeneNetwork. Genes. 2022; 13(4):614. https://doi.org/10.3390/genes13040614
Chicago/Turabian StyleChunduri, Alisha, Pamela M. Watson, and David G. Ashbrook. 2022. "New Insights on Gene by Environmental Effects of Drugs of Abuse in Animal Models Using GeneNetwork" Genes 13, no. 4: 614. https://doi.org/10.3390/genes13040614
APA StyleChunduri, A., Watson, P. M., & Ashbrook, D. G. (2022). New Insights on Gene by Environmental Effects of Drugs of Abuse in Animal Models Using GeneNetwork. Genes, 13(4), 614. https://doi.org/10.3390/genes13040614