
Osteogenesis Imperfecta/Ehlers–Danlos Overlap Syndrome and Neuroblastoma—Case Report and Review of Literature


 ,
,  , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Case Description
2.3. Samples
2.4. Next Generation Sequencing
3. Results
4. Discussion and Review of Literature
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lin, Z.; Zeng, J.; Wang, X. Compound phenotype of osteogenesis imperfecta and Ehlers Danlos syndrome caused by a combined mutations in COL1A1 and COL5A1. Biosci. Rep. 2019, 39, BSR20181409. [Google Scholar] [CrossRef]
- Van Dijk, F.S.; Sillence, D.O. Osteogenesis imperfecta: Clinical diagnosis, nomenclature, and severity assessment. Am. J. Med. Genet. 2014, 164, 1470–1481. [Google Scholar] [CrossRef]
- Marini, J.C.; Blissett, A.R. New genes in bone development: What’s the new in osteogenesis imperfecta. J. Clin. Endocrinol. Metab. 2013, 98, 3095–3103. [Google Scholar] [CrossRef]
- Marini, J.C.; Forlino, A.; Cabral, W.A.; Barnes, A.M.; San Antonio, J.D.; Milgrom, S.; Hyland, J.C.; Körkkö, J.; Prockop, D.J.; De Paepe, A.; et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: Regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum. Mutat. 2007, 28, 209–221. [Google Scholar] [CrossRef]
- Mortier, G.R.; Cohn, D.H.; Cormier-Daire, V.; Hall, C.; Krakow, D.; Mundlos, S.; Nishimura, G.; Robertson, S.; Sangiorgi, L.; Savarirayan, R.; et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. 2019, 179, 2393–2419. [Google Scholar] [CrossRef]
- Shapiro, F.; Maguire, K.; Swami, S.; Zhu, H.; Flynn, E.; Wang, J.; Wu, J.Y. Histopathology of osteogenesis imperfecta bone. Supramolecular assessment of cells and matrices in the context of woven and lamellar bone formation using light, polarization and ultrastructural microscopy. Bone Rep. 2020, 14, 100734. [Google Scholar] [CrossRef]
- Byers, P.H.; Krakow, D.; Nunes, M.E.; Pepin, M. Genetic evaluation of suspected osteogenesis imperfecta. Genet. Med. 2006, 8, 383–388. [Google Scholar] [CrossRef]
- Arvai, K.; Horvath, P.; Balla, B.; Tobias, B.; Katò, K.; Kirschner, G.; Klujber, V.; Lakatos, P.; Kosa, P. Next-generation sequencing of common osteogenesis imperfecta-related genes in clinical practice. Sci. Rep. 2016, 6, 28417. [Google Scholar] [CrossRef]
- Calzavara Pinton, P.; Ritelli, M. Delineation of Ehlers–Danlos syndrome phenotype due to the mutation (c.934C>T, p.Arg312Cys) in COL1A1: Report on a three-generation family without cardiovascular events, and literature review. Am. J. Med. Genet. 2017, 173, 524–530. [Google Scholar]
- Byers, P.H.; Duvic, M.; Atkinson, M.; Robinow, M.; Smith, L.T.; Krane, S.M.; Greally, M.T.; Ludman, M.; Matalon, R.; Pauker, S.; et al. Ehlers–Danlos syndrome type VIIA and VIIB result from splice-junction mutations or genomic deletions that involve exon 6 in the COL1A1 and COL1A2 genes of type I collagen. Am. J. Med. Genet. 1997, 72, 94–105. [Google Scholar] [CrossRef]
- Symoens, S.; Steyaert, W.; Demuynck, L.; De Paepe, A.; Diderich, K.E.; Malfait, F.; Coucke, P.J. Tissue-specific mosaicism for a lethal osteogenesis imperfecta COL1A1 mutation causes mild OI/EDS overlap syndrome. Am. J. Med. Genet. 2017, 173, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers–Danlos syndromes. Am. J. Med. Genet. Part. C Semin Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [PubMed]
- Morlino, S.; Micale, L.; Ritelli, M.; Rohrbach, M.; Zoppi, N.; Vandersteen, A.; Mackay, S.; Agolini, E.; Cocciadiferro, D.; Sasaki, E.; et al. COL1-related overlap disorder: A novel connective tissue incoroporating the osteogenesis imperfecta/Ehlers Danlos syndrome overlap. Clin. Genet. 2020, 97, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Cole, W.G. Etiology and pathogenesis of heritable connective tissue diseases. J. Pediatric Orthop. 1993, 13, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Raff, M.L.; Craigen, W.J.; Smith, L.T.; Keene, D.R.; Byers, P.H. Partial COL1A2 gene duplication produces features of osteogenesis imperfecta and Ehlers-Danlos syndrome type VII. Hum. Genet. 2000, 106, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Symoens, S.; Goemans, N.; Gyftodimou, Y.; Holmberg, E.; López-González, V.; Mortier, G.; Nampoothiri, S.; Petersen, M.B.; de Paepe, A. Helical mutations in type I collagen that afect the processing of the amino-propeptide result in an Osteogenesis Imperfecta/Ehlers Danlos Syndrome overlap syndrome. Orphanet J. Rare Dis. 2013, 8, 78. [Google Scholar] [CrossRef]
- Binkovitz, L.A.; Henwood, M.J. Pediatric DXA: Technique and interpretation. Pediatric Radiol. 2007, 37, 21–31. [Google Scholar] [CrossRef]
- Lindahl, K.; Åström, E.; Rubin, C.J.; Grigelioniene, G.; Malmgren, B.; Ljunggren, Ö.; Kindmark, A. Genetic epidemiology, prevalence, and genotype-phenotype correlations in the Swedish population with osteogenesis imperfecta. Eur. J. Hum. Genet. 2015, 23, 1042–1050. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; ACMG Laboratory Quality Assurance Committee; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Zhytnik, L.; Maasalu, K.; Reimann, E.; Prans, E.; Kõks, S.; Märtson, A. Mutation analysis of COL1A1 and COL1A2 genes among Estonian osteogenesis imperfecta patients. Hum. Genet. 2017, 11, 19. [Google Scholar] [CrossRef]
- Cabral, W.A.; Makareeva, E.; Letocha, A.D.; Scribanu, N.; Fertala, A.; Steplewski, A.; Keene, D.R.; Persikov, A.V.; Leikin, S.; Marini, J.C. Y-position cysteine substitution in type I collagen (alpha1(I) R888C/p.R1066C) is associated with osteogenesis imperfecta/Ehlers Danlos syndrome phenotype. Hum. Mutat. 2007, 28, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, A.C.; Valler, D.; Wallis, S.; Pope, F.M. Homozigosity for a splice site mutation of the COL1A2 gene yields a non functional pro(α)2(I) chain and an EDS/OI clinical phenotype. J. Med. Genet. 2001, 38, 132–136. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bella, J.; Eaton, M.; Brodsky, B.; Berman, H.M. Crystal and molecular structure of a collagen-like peptide at 1.9 A resolution. Science 1994, 266, 75–81. [Google Scholar] [CrossRef]
- Long, C.G.; Braswell, E.; Zhu, D.; Apigo, J.; Baum, J.; Brodsky, B. Characterization of collagen-like peptides containing interruptions in the repeating Gly-X-Y sequence. Biochemistry 1993, 32, 11688–11695. [Google Scholar] [CrossRef]
- Shoulders, M.D.; Raines, R.T. Collagen structure and stability. Annu. Rev. Biochem. 2009, 78, 929–958. [Google Scholar] [CrossRef]
- Mottes, M.; Sangalli, A.; Valli, M.; Gomez Lira, M.; Tenni, R.; Buttitta, P.; Pignatti, P.F.; Cetta, G. Mild dominant osteogenesis imperfecta with intrafamilial variability: The cause is a serine for glycine α 1(I) 901 substitution in a type-I collagen gene. Hum. Genet. 1992, 89, 480–484. [Google Scholar] [CrossRef]
- Kaneko, H.; Kitoh, H.; Matsuura, T.; Masuda, A.; Ito, M.; Mottes, M.; Rauch, F.; Ishiguro, N.; Ohno, K. Hyperuricemia cosegregating with osteogenesis imperfecta is associated with a mutation in GPATCH8. Hum. Genet. 2011, 130, 671–863. [Google Scholar] [CrossRef]
- Ju, M.; Zhang, T.; Bai, X.; Ren, X.; Li, K.; Li, G. Analysis of type IV osteogenesis imperfecta caused by two mutations occurred simultaneously in COL1A1 gene in a Chinese child. Chin. J. Med. Genet. 2016, 33, 140–144. [Google Scholar] [CrossRef]
- Hartikka, H.; Kuurila, K.; Körkkö, J.; Kaitila, I.; Grénman, R.; Pynnönen, S.; Hyland, J.C.; Ala-Kokko, L. Lack of correlation between the type of COL1A1 or COL1A2 mutation and hearing loss in osteogenesis imperfecta patients. Hum. Mutat. 2004, 27, 147–154. [Google Scholar] [CrossRef]
- Moraes, M.V.; Milanez, M.; Almada, B.V.; Sipolatti, V.; Rebouças, M.R.; Nunes, V.R.; Akel, A.N.; Zatz, M.; Errera, F.I.; Louro, I.D.; et al. Variable expressivity of osteogenesis imperfecta in a Brazilian family due to p.G1079S mutation in the COL1A1 gene. Genet. Mol. Res. 2012, 11, 3246–3255. [Google Scholar] [CrossRef]
- Roschger, P.; Fratzl-Zelman, N.; Misof, B.M.; Glorieux, F.H.; Klaushofer, K.; Rauch, F. Evidence that abnormal high bone mineralization in growing children with osteogenesis imperfecta is not associated with specific collagen mutations. Calcif. Tissue Int. 2008, 82, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Bardai, G.; Moffatt, P.; Glorieux, F.H.; Rauch, F. DNA sequence analysis in 598 individuals with a clinical diagnosis of osteogenesis imperfecta: Diagnostic yield and mutation spectrum. Osteoporos. Int. 2016, 27, 3607–3613. [Google Scholar] [CrossRef] [PubMed]
- Andersson, K.; Dahllöf, G.; Lindahl, K.; Kindmark, A.; Grigelioniene, G.; Åström, E.; Malmgren, B. Mutations in COL1A1 and COL1A2 and dental aberrations in children and adolescents with osteogenesis imperfecta-A retrospective cohort study. PLoS ONE 2017, 12, e0176466. [Google Scholar] [CrossRef]
- Budsamongkol, T.; Intarak, N.; Theerapanon, T.; Yodsanga, S.; Porntaveetus, T.; Shoterlersuk, V. A novel mutation in COL1A2 leads to osteogenesis imperfecta/Ehlers Danlos overlap syndrome with brachydactyly. Genet. Dis. 2019, 6, 138–146. [Google Scholar] [CrossRef]
- Marom, R.; Rabenhorst, B.M.; Morello, R. Osteogenesis imperfecta: An update on clinical features and therapies. Eur. J. 2020, 183, R95–R106. [Google Scholar] [CrossRef]
- Zhang, Z.L.; Zhang, H.; Ke, Y.H.; Yue, H.; Xiao, W.J.; Yu, J.B.; Gu, J.M.; Hu, W.W.; Wang, C.; He, J.W.; et al. The identification of novel mutations in COL1A1, COL1A2, and LEPRE1 genes in Chinese patients with osteogenesis imperfecta. J. Bone Metab. 2012, 30, 69–77. [Google Scholar] [CrossRef]
- Feshchenko, S.; Brinckmann, J.; Lehmann, H.W.; Koch, H.G.; Muller, P.K.; Kugler, S. Identification of a new heterozygous point mutation in the COL1A2 gene leading to skipping of exon 9 in a patient with joint laxity, hyperextensibility of skin and blue sclerae. Mutation in brief no. 166. Hum. Mutat. 1998, 12, 138. [Google Scholar] [CrossRef]
- Wu, Q.Q.; Chen, Q. Mechanoregulation of chondrocyte proliferation, maturation and hypertrophy: Ion-channel dependent transduction of matrix deformation signals. Exp. Cell Res. 2000, 256, 383–391. [Google Scholar] [CrossRef]
- Scheiber, A.L.; Guess, A.J.; Kaito, T.; Abzurg, J.M.; Enomoto- Iwamoto, M.; Leikin, S.; Iwamoto, M.; Ostsurus, S. Endoplasmic reticulum stress is induced in growth plate hypertrophic chondrocytes in G610C mouse model of Osteogenesis imperfecta. Biochem. Biophys. Res. Commun. 2019, 509, 235–240. [Google Scholar] [CrossRef]
- Zeitlin, L.; Rauch, F.; Plotkin, H.; Glorieux, F.H. Height and weight development during four years of therapy with cyclical intravenous pamidronate in children and adolescents with osteogenesis imperfecta types I.; III, and IV. Pediatric 2003, 111, 1030–1036. [Google Scholar] [CrossRef]
- Hoyer-Kuhn, H.; Hobing, L.; Cassens, J.; Schoenau, E.; Semler, O. Children with severe Osteogenesis imperfecta and short stature present on average with normal IGF-1 and IGFBP-3 levels. J. Pediatric Endocrinol. Metab. 2016, 29, 813–818. [Google Scholar] [CrossRef]
- Boum, W.F.; Cao, D.; Hesse, V.; Fricke-Otto, S.; Ross, J.L.; Jones, C.; Quigley, C.A.; Binder, G. Height gains in response to growth hormone treatment to final height are similar in patients with SHOX deficiency and Turner Syndrome. Horm. Res. Paediatr. 2009, 71, 167–172. [Google Scholar]
- Kumps, C.; Campos-Xavier, B.; Hilhorst-Hofstee, Y.; Marcelis, C.; Kraenzlin, M.; Fleischer, N.; Unger, S.; Superti-Furga, A. The Connective Tissue Disorder Associated with Recessive Variants in the SLC39A13 Zinc Transporter Gene (Spondylo-Dysplastic Ehlers-Danlos Syndrome Type 3): Insights from Four Novel Patients and Follow-Up on Two Original Cases. Genes 2020, 11, 420. [Google Scholar] [CrossRef]
- Giunta, C.; Elcioglu, N.H.; Albrecht, B.; Eich, G.; Chambaz, C.; Janecke, A.R.; Yeowell, H.; Weis, M.; Eyre, D.R.; Kraenzlin, M.; et al. Spondylocheiro dysplastic form of the Ehlers-Danlos syndrome—An autosomal-recessive entity caused by mutations in the zinc transporter gene SLC39A13. Am. J. Hum. Genet. 2008, 82, 1290–1305. [Google Scholar] [CrossRef]
- Writing Group for Practice Guidelines for Diagnosis and Treatment of Genetic Diseases Medical Genetics Branch of Chinese Medical Association; Li, C.; Xie, B.; Shen, Y.; Luo, F. Clinical practice guidelines for Prader-Willi syndrome. Chin. J. Med. Genet. 2020, 37, 318–323. [Google Scholar] [CrossRef]
- Mizrak, D.; Alkan, A.; Erdogdu, B.; Utkan, G. Osteogenesis imperfecta, pseudoachalasia, and gastric cancer. Case Rep. Gastrointest. Med. 2015, 2015, 685459. [Google Scholar] [CrossRef]
- Rosenstock, H.A. Osteogenesis imperfecta: Biochemical cancer resistance? Family pedigree and review of literature. Tex Med. 1970, 66, 44–47. [Google Scholar]
- Klenerman, L.; Ockenden, B.G.; Townsend, A.C. Osteosarcoma occurring in osteogenesis imperfecta. Report of two cases. Bone Jt. J. 1967, 49, 314–323. [Google Scholar] [CrossRef][Green Version]
- Takahashi, S.; Okada, K.; Nagasawa, H.; Shimada, Y.; Sakamoto, H.; Itoi, E. Osteosarcoma occurring in osteogenesis imperfecta. Virchows Arch. 2004, 444, 454–458. [Google Scholar] [CrossRef]
- Beuzeboc, P.; Pierga, J.Y.; Stoppalyonnet, D.; Etienne, M.C.; Milano, G.; Pouillart, P. Severe 5-fluorouracil toxicity possibly secondary to dihydropyrimidine dehydrogenase deficiency in a breast cancer patient with osteogenesis imperfecta. Eur. J. Cancer 1996, 32, 369–370. [Google Scholar] [CrossRef]
- Augustin, G.; Jelincic, Z.; Majerovic, M.; Stefancic, L. Carcinoma of left colon presenting as mechanical obstruction in a patient with osteogensesis imperfecta type III. J. Inherit. Metab. Dis. 2007, 30, 109–110. [Google Scholar] [CrossRef][Green Version]
- Nishida, T.; Oda, T.; Sugiyama, T.; Izumi, S.; Yakushiji, M. Concurrent ovarian serous carcinoma and osteogenesis imperfecta. Arch. Gynecol. Obstet. 1993, 253, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, S.; Nakahara, M.; Kido, T.; Imabun, S.; Akamatsu, H.; Kurozumi, K.; Kimura, I.; Fukui, S.; Nakao, K.; Tsujimoto, M. A case of advanced colon cancer associated with osteogenesis imperfecta. J. Gastrointest. Surg. 2001, 34, 1466–1470. [Google Scholar]
- Bohler, F.K.; Rhomberg, W. Osteogenesis imperfecta and breast carcinoma. A case study of radiobiological interest. Strahelnther. Onkol. 1994, 170, 665–667. [Google Scholar]
- Taira, F.; Shimizu, H.; Kosaka, T.; Saito, M.; Kasumi, F. An osteogenesis imperfecta case with breast cancer. Breast Cancer 2014, 21, 769–773. [Google Scholar] [CrossRef]
- Monclair, T.; Brodeur, G.M.; Ambros, P.F.; Brisse, H.J.; Cecchetto, G.; Holmes, K.; Kaneko, M.; London, W.B.; Matthay, K.K.; Nuchtern, J.G.; et al. The International Neuroblastoma Risk Group (INRG) staging system: An INRG task force report. J. Clin. Oncol. 2009, 27, 298–303. [Google Scholar] [CrossRef]
- Tonini, G.P.; Capasso, M. Genetic predisposition and chromosome instability in neuroblastoma. Cancer Metastasis Rev. 2020, 39, 275–285. [Google Scholar] [CrossRef]
- Berthold, F.; Rosswog, C.; Christiansen, H.; Frühwald, M.; Hemstedt, N.; Klingebiel, T.; Fröhlich, B.; Schilling, F.H.; Schmid, I.; Simon, T.; et al. Clinical and molecular characterization of patients with stage 4(M) neuroblastoma aged less than 18 months without MYCN amplification. Pediatric Blood Cancer 2021, 68, 290. [Google Scholar] [CrossRef]
- De Bernardi, B.; Gerrard, M.; Boni, L.; Rubie, H.; Cañete, A.; Di Cataldo, A.; Castel, V.; de Lacerda, A.F.; Ladenstein, R.; Ruud, E.; et al. Excellent outcome with reduced treatment for infants with disseminated neuroblastoma without MYCN gene amplification. J. Clin. Oncol. 2009, 27, 1034–1040. [Google Scholar] [CrossRef]
- Lasorsa, V.A.; Formicola, D.; Pignataro, P.; Cimmino, F.; Calabrese, F.M.; Mora, J.; Esposito, M.R.; Pantile, M.; Zanon, C.; De Mariano, M.; et al. Exome and deep sequencing of clinically aggressive neuroblastoma reveal somatic mutations that affect key pathways involved in cancer progression. Oncotarget 2016, 7, 21840–21852. [Google Scholar] [CrossRef]
- Gartlgruber, M.; Sharma, A.K.; Quintero, A.; Dreidax, D.; Jansky, S.; Park, Y.-G.; Kreth, S.; Meder, J.; Doncevic, D.; Saary, P.; et al. Super enhancers define regulatory subtypes and cell identity in neuroblastoma. Nat. Cancer 2020, 2, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Song, Z.; Hu, B.; Chen, Z.; Chen, F.; Cao, C. MicroRNA642a5p inhibits colon cancer cell migration and invasion by targeting collagen type I α1. Oncol. Rep. 2021, 45, 933–944. [Google Scholar] [CrossRef]
- Guo, Y.; Lu, G.; Mao, H.; Zhou, S.; Tong, X.; Wu, J.; Sun, Q.; Xu, H.; Fang, F. miR-133b Suppresses Invasion and Migration of Gastric Cancer Cells via the COL1A1/TGF-β Axis. OncoTargets Ther. 2020, 13, 7985–7995. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sun, H.; Zhao, Y.; Zhang, J.; Xiong, G.; Cui, Y.; Lei, C. CircRNA circ_0004370 promotes cell proliferation, migration, and invasion and inhibits cell apoptosis of esophageal cancer via miR-1301-3p/COL1A1 axis. Open Med. 2021, 16, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, J.; Wang, C.; Xia, L.; Xu, J.; Xie, X.; Lu, W. Microenvironment remodeled by tumor and stromal cells elevates fibroblast-derived COL1A1 and facilitates ovarian cancer metastasis. Exp. Cell Res. 2020, 394, 112153. [Google Scholar] [CrossRef]


{kind=link}
| References | No. of Case Described | Gene | Nucleotide Change | De Novo (DN)/Familial (F) Case | Blue Sclerae | Hyper Extensible Skin | Positive Beighton Score | History of Fractures | Altered Bone Density | Short Stature | Cardiac Valvular Defects | Easy Bruising | Hearing Loss | Joint Pain | Other Features |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Morlino et al., 2020 | 21 | COL1A1 (6 cases) | c.2073delT | F | + | + | + | + | - | - | + | - | - | + | Piezogenic papules |
| c.1243C>T | F | + | + | + | + | + | - | + | - | - | + | Flatfeet | |||
| c.670G>A | F | + | + | + | + | - | + | NA | - | - | + | Muscle ruptures | |||
| c.581G>C | F | + | + | + | - | - | - | NA | - | + | + | Microcornea progressive scoliosis | |||
| c.326G>A | F | + | + | + | - | - | - | NA | - | - | + | Dystrophic scars | |||
| COL1A2 (15 cases) | c.577G>A | F | + | +/− | + | + | + | - | + | + | + | + | Chronic periodontitis, neonatal hypotonia | ||
| c.432 + 5G>A | + | - | + | + | NA | - | - | - | - | - | --- | ||||
| c.335G>T | + | - | + | + | - | - | NA | - | - | + | Flat feet, progressive scoliosis | ||||
| c.197G>A | + | + | + | + | - | - | NA | - | - | + | Dental crowding, high arched palate | ||||
| c.432+ + 4_432 + 7delAGTA | F | + | + | + | - | - | - | + | + | - | + | Flatfeet | |||
| c133G>T | F | - | + | + | + | - | - | - | + | - | - | Myopia, gingival fragility | |||
| c.316G>A | F | + | - | + | + | - | + | + | + | - | + | Myopia, high arched palate | |||
| c.2755G>A | DN | + | - | + | + | - | + | NA | + | - | + | Flatfeet | |||
| Budsamongkol et al., 2019 | 1 | COL1A2 | c.3296G>A | DN | + | + | + | + | + | + | NA | - | NA | - | Brachydactyly, malocclusion dentinogenesis imperfecta, skeletal deformities |
| Lin et al., 2019 | 1 | COL1A1 COL5A1 | c.2010delT c.5335A>G | F | + | - | + | + | NA | NA | NA | + | - | - | Prominent ears, atrophic scarring |
| Mackenroth et al., 2016 | 1 | COL1A1 TNXB | c.4006-1G>A c.7774G>A c.3637G>A | F | - | - | + | + | + | - | - | - | - | - | Severe muscular weakness, abnormally shaped vertebra |
| Malfait et al., 2013 | 7 | COL1A1 | c.563G>A | DN | + | + | - | + | + | + | - | + | NA | NA | Atrial septum defect, muscular hypotonia, arterial rupture |
| c.607G>T | DN | + | + | + | + | + | + | - | + | NA | NA | Aortic dilation, inguinal hernia joint dislocation, kyphoscoliosis | |||
| COL1A2 | c.324+4delA | DN | + | + | + | + | + | + | + | + | NA | NA | Muscular hypotonia, joint dislocation | ||
| c.587G>T | F | + | + | NA | - | + | NA | - | + | NA | NA | Muscular hypotonia, intracranial bleeding | |||
| c.432 + 4_432 + 7delAGTA | DN | + | + | + | + | + | + | - | + | NA | NA | Joint dislocations | |||
| c.587G>T | DN | + | + | + | + | + | + | - | + | NA | NA | Muscular hypotonia | |||
| c.693+5G>A | DN | + | + | NA | + | + | - | NA | + | NA | NA | Pes planus, mild bowing of tibia and fibula | |||
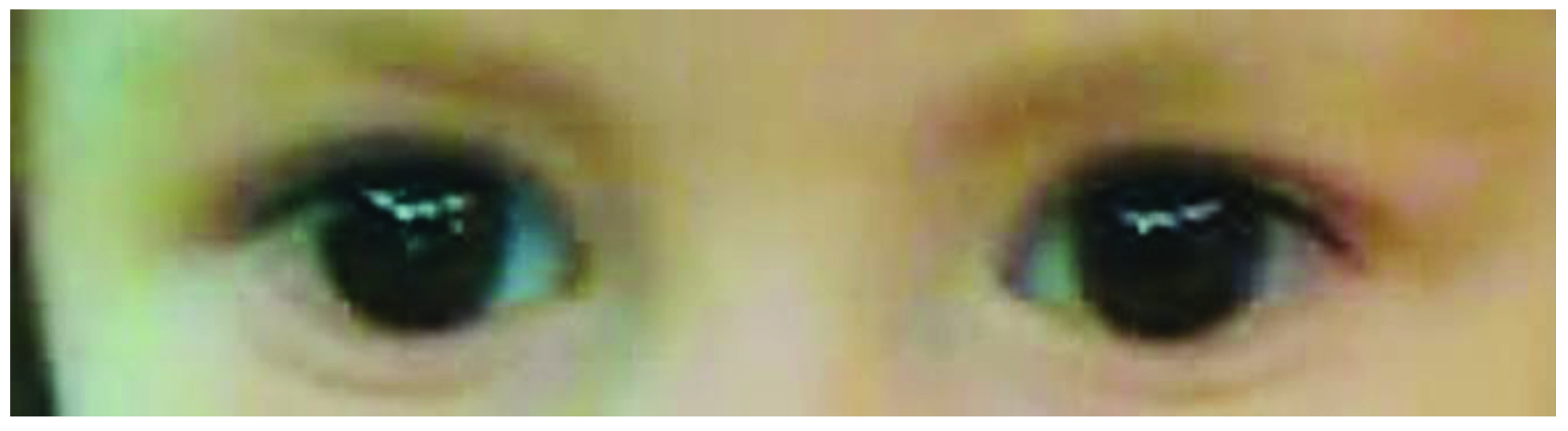
| Present report | 1 | COL1A1 | c.3235G>A | DN | + | + | + | - | + | + | - | - | - | - | Changes in tooth enamel, history of neuroblastoma, scoliosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morabito, L.A.; Allegri, A.E.M.; Capra, A.P.; Capasso, M.; Capra, V.; Garaventa, A.; Maghnie, M.; Briuglia, S.; Wasniewska, M.G. Osteogenesis Imperfecta/Ehlers–Danlos Overlap Syndrome and Neuroblastoma—Case Report and Review of Literature. Genes 2022, 13, 581. https://doi.org/10.3390/genes13040581
Morabito LA, Allegri AEM, Capra AP, Capasso M, Capra V, Garaventa A, Maghnie M, Briuglia S, Wasniewska MG. Osteogenesis Imperfecta/Ehlers–Danlos Overlap Syndrome and Neuroblastoma—Case Report and Review of Literature. Genes. 2022; 13(4):581. https://doi.org/10.3390/genes13040581
Chicago/Turabian StyleMorabito, Letteria Anna, Anna Elsa Maria Allegri, Anna Paola Capra, Mario Capasso, Valeria Capra, Alberto Garaventa, Mohamad Maghnie, Silvana Briuglia, and Malgorzata Gabriela Wasniewska. 2022. "Osteogenesis Imperfecta/Ehlers–Danlos Overlap Syndrome and Neuroblastoma—Case Report and Review of Literature" Genes 13, no. 4: 581. https://doi.org/10.3390/genes13040581
APA StyleMorabito, L. A., Allegri, A. E. M., Capra, A. P., Capasso, M., Capra, V., Garaventa, A., Maghnie, M., Briuglia, S., & Wasniewska, M. G. (2022). Osteogenesis Imperfecta/Ehlers–Danlos Overlap Syndrome and Neuroblastoma—Case Report and Review of Literature. Genes, 13(4), 581. https://doi.org/10.3390/genes13040581