
RNA Sequencing of Whole Blood Defines the Signature of High Intensity Exercise at Altitude in Elite Speed Skaters

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects and Study Protocol
2.2. Blood Sample Collection and RNA Isolation
2.3. Library Preparation and Illumina RNA Sequencing
2.4. Alignment and Quantification
2.5. Differential Expression and Pathway Enrichment Analysis
2.6. Public Single Cell RNA-Seq Dataset Processing
2.7. Microarray Dataset Reanalysis
3. Results
3.1. Blood Panel and Physiological Measurements
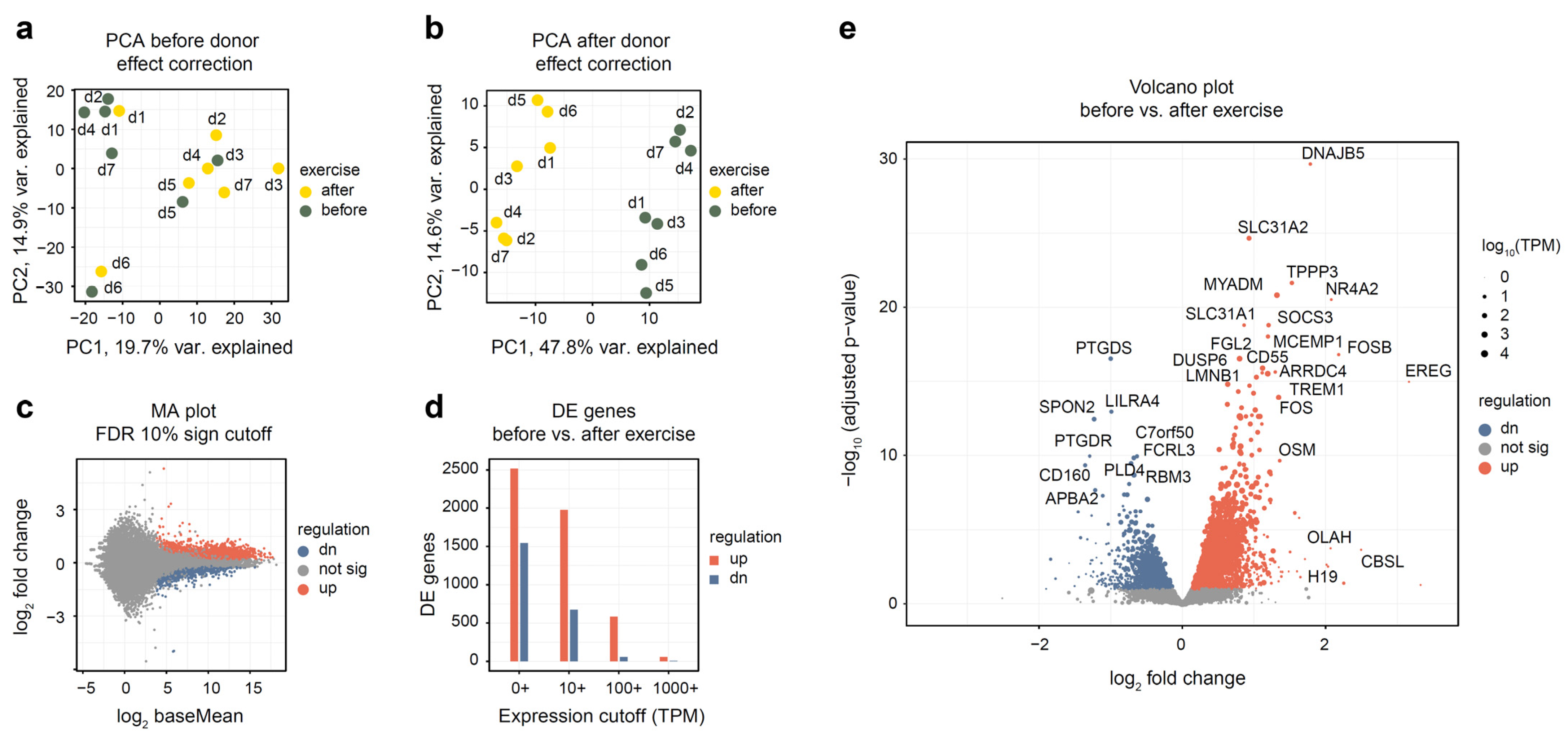
3.2. Differential Gene Expression Analysis
3.3. Pathway and Functional Category Analysis
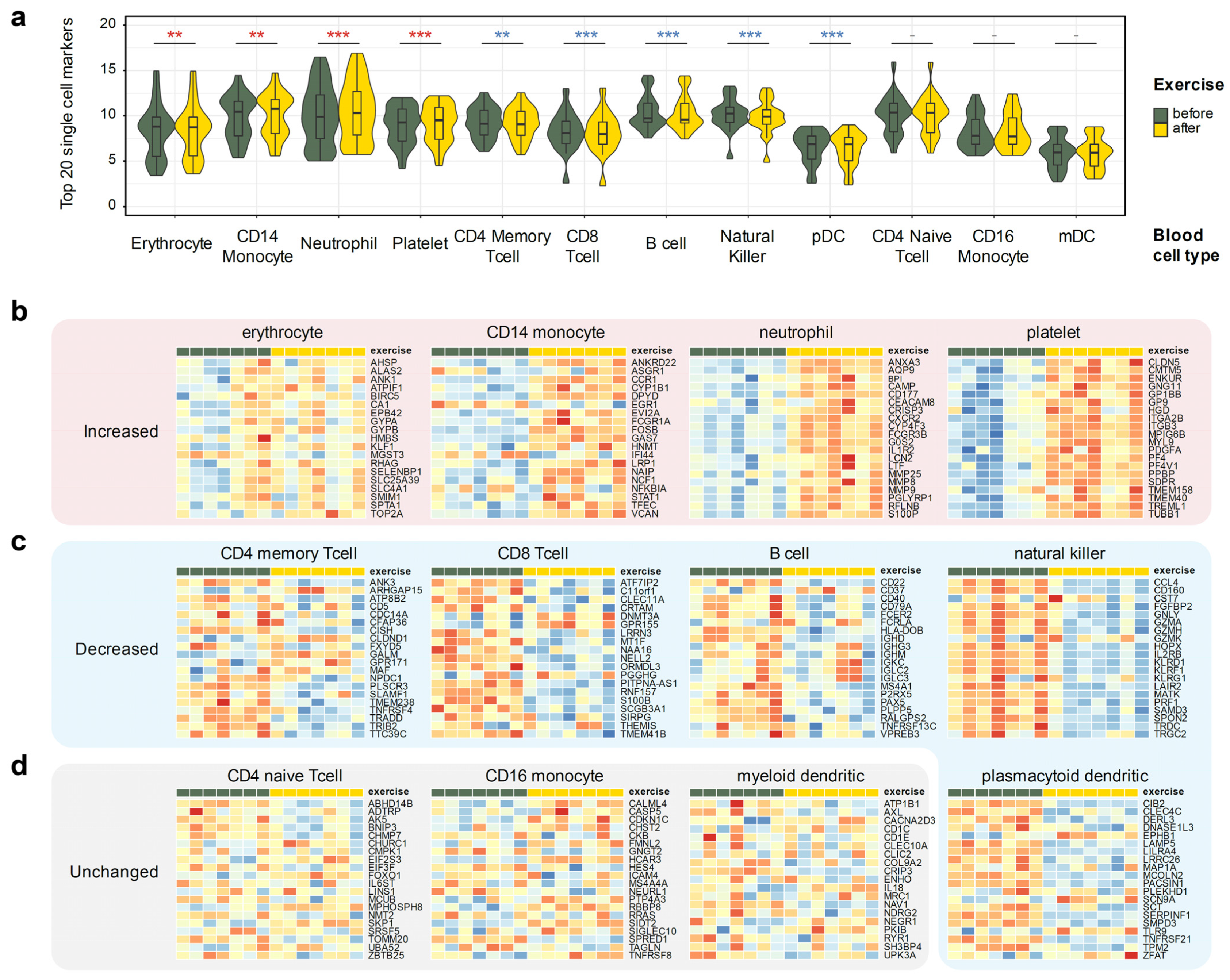
3.4. Analysis of Cell Type Composition Changes Based on Expression Signatures
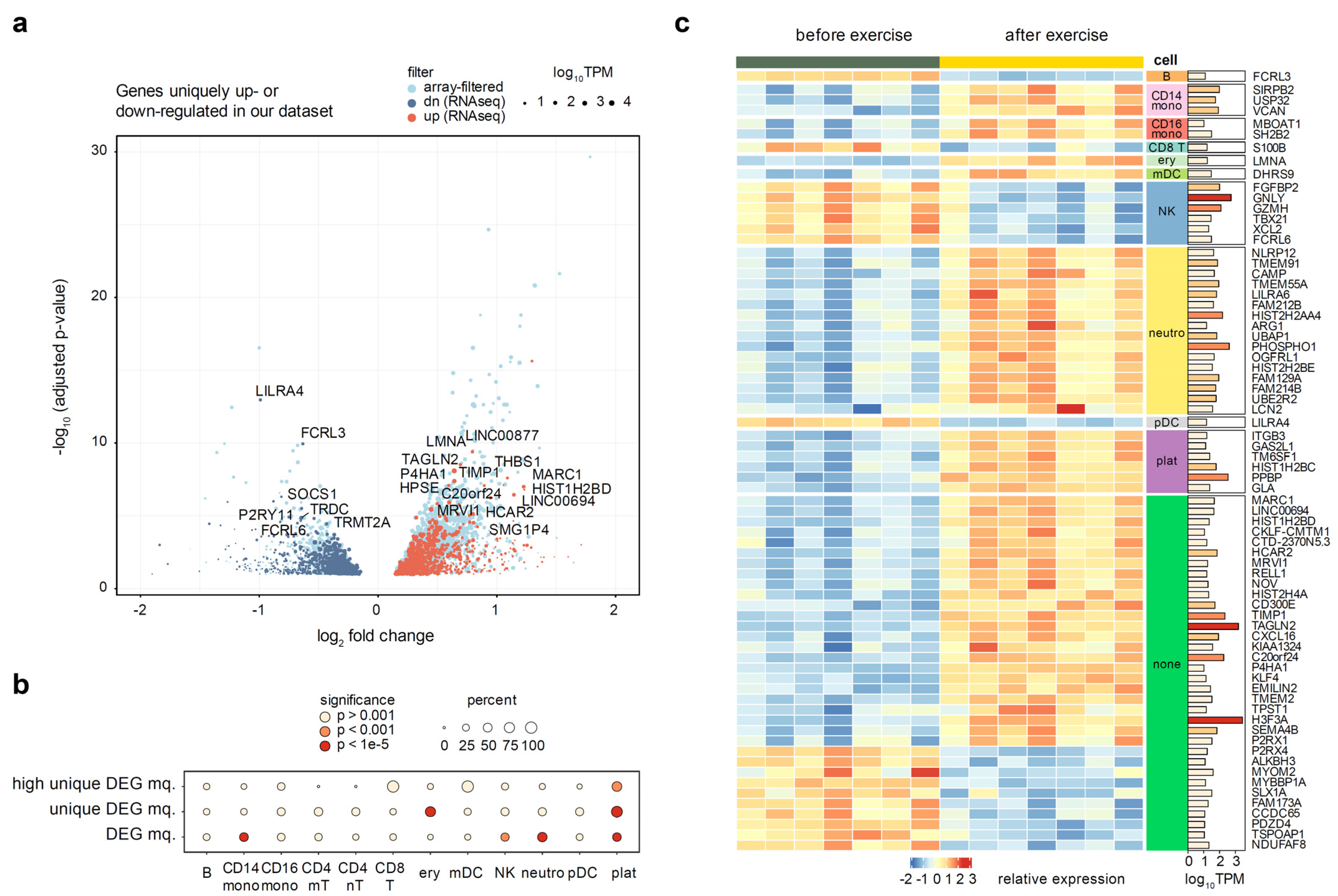
3.5. Context of Other Exercise Expression Datasets
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gjevestad, G.O.; Holven, K.B.; Ulven, S.M. Effects of Exercise on Gene Expression of Inflammatory Markers in Human Peripheral Blood Cells: A Systematic Review. Curr. Cardiovasc. Risk Rep. 2015, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Nieman, D.C.; Pence, B.D. Exercise Immunology: Future Directions. J. Sport Health Sci. 2020, 9, 432–445. [Google Scholar] [CrossRef]
- Nieman, D.C.; Wentz, L.M. The Compelling Link between Physical Activity and the Body’s Defense System. J. Sport Health Sci. 2019, 8, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Larrabee, R.C. Leucocytosis after Violent Exercise. J. Med. Res. 1902, 7, 76–82. [Google Scholar] [PubMed]
- Bull, F.C.; Al-Ansari, S.S.; Biddle, S.; Borodulin, K.; Buman, M.P.; Cardon, G.; Carty, C.; Chaput, J.-P.; Chastin, S.; Chou, R.; et al. World Health Organization 2020 Guidelines on Physical Activity and Sedentary Behaviour. Br. J. Sports Med. 2020, 54, 1451–1462. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Wang, C.; Gong, B.; Bushel, P.R.; Thierry-Mieg, J.; Thierry-Mieg, D.; Xu, J.; Fang, H.; Hong, H.; Shen, J.; Su, Z.; et al. The Concordance between RNA-Seq and Microarray Data Depends on Chemical Treatment and Transcript Abundance. Nat. Biotechnol. 2014, 32, 926–932. [Google Scholar] [CrossRef]
- Timp, W.; Timp, G. Beyond Mass Spectrometry, the next Step in Proteomics. Sci. Adv. 2020, 6, eaax8978. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Q.; Xu, Z.; Dou, J. Mass Spectrometry-Based Metabolomics in Health and Medical Science: A Systematic Review. RSC Adv. 2020, 10, 3092–3104. [Google Scholar] [CrossRef]
- Fabre, O.; Ingerslev, L.R.; Garde, C.; Donkin, I.; Simar, D.; Barrès, R. Exercise Training Alters the Genomic Response to Acute Exercise in Human Adipose Tissue. Epigenomics 2018, 10, 1033–1050. [Google Scholar] [CrossRef]
- Gustafsson, T.; Puntschart, A.; Kaijser, L.; Jansson, E.; Sundberg, C.J. Exercise-Induced Expression of Angiogenesis-Related Transcription and Growth Factors in Human Skeletal Muscle. Am. J. Physiol. Heart Circ. Physiol. 1999, 276, H679–H685. [Google Scholar] [CrossRef]
- Hawley, J.A.; Lundby, C.; Cotter, J.D.; Burke, L.M. Maximizing Cellular Adaptation to Endurance Exercise in Skeletal Muscle. Cell Metab. 2018, 27, 962–976. [Google Scholar] [CrossRef] [PubMed]
- Terry, E.E.; Zhang, X.; Hoffmann, C.; Hughes, L.D.; Lewis, S.A.; Li, J.; Wallace, M.J.; Riley, L.A.; Douglas, C.M.; Gutierrez-Monreal, M.A.; et al. Transcriptional Profiling Reveals Extraordinary Diversity among Skeletal Muscle Tissues. eLife 2018, 7, e34613. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.D.; Farrell, L.; Wood, M.J.; Martinovic, M.; Arany, Z.; Rowe, G.C.; Souza, A.; Cheng, S.; McCabe, E.L.; Yang, E.; et al. Metabolic Signatures of Exercise in Human Plasma. Sci. Transl. Med. 2010, 2, 33ra37. [Google Scholar] [CrossRef] [PubMed]
- Donohue, D.E.; Gautam, A.; Miller, S.-A.; Srinivasan, S.; Abu-Amara, D.; Campbell, R.; Marmar, C.R.; Hammamieh, R.; Jett, M. Gene Expression Profiling of Whole Blood: A Comparative Assessment of RNA-Stabilizing Collection Methods. PLoS ONE 2019, 14, e0223065. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Ferreira, A.-M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE Reference Annotation for the Human and Mouse Genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-Performance Genomics Data Visualization and Exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The Sva Package for Removing Batch Effects and Other Unwanted Variation in High-Throughput Experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular Signatures Database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Korotkevich, G.; Sukhov, V.; Budin, N.; Shpak, B.; Artyomov, M.N.; Sergushichev, A. Fast Gene Set Enrichment Analysis. bioRxiv 2016, 060012. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Meltzer, P.S. GEOquery: A Bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Horscroft, J.A.; Kotwica, A.O.; Laner, V.; West, J.A.; Hennis, P.J.; Levett, D.Z.H.; Howard, D.J.; Fernandez, B.O.; Burgess, S.L.; Ament, Z.; et al. Metabolic Basis to Sherpa Altitude Adaptation. Proc. Natl. Acad. Sci. USA 2017, 114, 6382–6387. [Google Scholar] [CrossRef]
- Moore, L.G. Measuring High-Altitude Adaptation. J. Appl. Physiol. 2017, 123, 1371–1385. [Google Scholar] [CrossRef]
- Cai, Q.; Medeiros, L.J.; Xu, X.; Young, K.H. MYC -Driven Aggressive B-Cell Lymphomas: Biology, Entity, Differential Diagnosis and Clinical Management. Oncotarget 2015, 6, 38591–38616. [Google Scholar] [CrossRef] [PubMed]
- Mooren, F.C.; Blöming, D.; Lechtermann, A.; Lerch, M.M.; Völker, K. Lymphocyte Apoptosis after Exhaustive and Moderate Exercise. J. Appl. Physiol. 2002, 93, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Liu, M.; Zhang, Y.; Wang, B.; Zhu, C.; Wang, C.; Li, Q.; Huo, Y.; Guo, J.; Xu, C.; et al. Single-Cell Transcriptomic Landscape of Human Blood Cells. Natl. Sci. Rev. 2021, 8, nwaa180. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively Parallel Digital Transcriptional Profiling of Single Cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [PubMed]
- Büttner, P.; Mosig, S.; Lechtermann, A.; Funke, H.; Mooren, F.C. Exercise Affects the Gene Expression Profiles of Human White Blood Cells. J. Appl. Physiol. 2007, 102, 26–36. [Google Scholar] [CrossRef]
- Connolly, P.H.; Caiozzo, V.J.; Zaldivar, F.; Nemet, D.; Larson, J.; Hung, S.; Heck, J.D.; Hatfield, G.W.; Cooper, D.M. Effects of Exercise on Gene Expression in Human Peripheral Blood Mononuclear Cells. J. Appl. Physiol. 2004, 97, 1461–1469. [Google Scholar] [CrossRef]
- Mukherjee, K.; Edgett, B.A.; Burrows, H.W.; Castro, C.; Griffin, J.L.; Schwertani, A.G.; Gurd, B.J.; Funk, C.D. Whole Blood Transcriptomics and Urinary Metabolomics to Define Adaptive Biochemical Pathways of High-Intensity Exercise in 50-60 Year Old Masters Athletes. PLoS ONE 2014, 9, e92031. [Google Scholar] [CrossRef]
- Nakamura, S.; Kobayashi, M.; Sugino, T.; Kajimoto, O.; Matoba, R.; Matsubara, K. Effect of Exercise on Gene Expression Profile in Unfractionated Peripheral Blood Leukocytes. Biochem. Biophys. Res. Commun. 2010, 391, 846–851. [Google Scholar] [CrossRef]
- Radom-Aizik, S.; Zaldivar, F.; Leu, S.-Y.; Cooper, D.M. Brief Bout of Exercise Alters Gene Expression in Peripheral Blood Mononuclear Cells of Early- and Late-Pubertal Males. Pediatr. Res. 2009, 65, 447–452. [Google Scholar] [CrossRef]
- Radom-Aizik, S.; Zaldivar, F.; Leu, S.-Y.; Cooper, D.M. A Brief Bout of Exercise Alters Gene Expression and Distinct Gene Pathways in Peripheral Blood Mononuclear Cells of Early- and Late-Pubertal Females. J. Appl. Physiol. 2009, 107, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Tonevitsky, A.G.; Maltseva, D.V.; Abbasi, A.; Samatov, T.R.; Sakharov, D.A.; Shkurnikov, M.U.; Lebedev, A.E.; Galatenko, V.V.; Grigoriev, A.I.; Northoff, H. Dynamically Regulated MiRNA-MRNA Networks Revealed by Exercise. BMC Physiol. 2013, 13, 9. [Google Scholar] [CrossRef]
- Sakharov, D.A.; Maltseva, D.V.; Riabenko, E.A.; Shkurnikov, M.U.; Northoff, H.; Tonevitsky, A.G.; Grigoriev, A.I. Passing the Anaerobic Threshold Is Associated with Substantial Changes in the Gene Expression Profile in White Blood Cells. Eur. J. Appl. Physiol. 2012, 112, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Kubitza, C.; Bittner, F.; Ginsel, C.; Havemeyer, A.; Clement, B.; Scheidig, A.J. Crystal Structure of Human MARC1 Reveals Its Exceptional Position among Eukaryotic Molybdenum Enzymes. Proc. Natl. Acad. Sci. USA 2018, 115, 11958–11963. [Google Scholar] [CrossRef]
- Kotthaus, J.; Wahl, B.; Havemeyer, A.; Kotthaus, J.; Schade, D.; Garbe-Schönberg, D.; Mendel, R.; Bittner, F.; Clement, B. Reduction of Nω-Hydroxy-L-Arginine by the Mitochondrial Amidoxime Reducing Component (MARC). Biochem. J. 2011, 433, 383–391. [Google Scholar] [CrossRef]
- Loscalzo, J.; Welch, G. Nitric Oxide and Its Role in the Cardiovascular System. Prog. Cardiovasc. Dis. 1995, 38, 87–104. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, X.; Noviana, M.; Hou, M. Nitric Oxide in Red Blood Cell Adaptation to Hypoxia. Acta Biochim. Biophys. Sin. 2018, 50, 621–634. [Google Scholar] [CrossRef]
- Thul, P.J.; Lindskog, C. The Human Protein Atlas: A Spatial Map of the Human Proteome: The Human Protein Atlas. Protein Sci. 2018, 27, 233–244. [Google Scholar] [CrossRef]
- Huang, N.-J.; Lin, Y.-C.; Lin, C.-Y.; Pishesha, N.; Lewis, C.A.; Freinkman, E.; Farquharson, C.; Millán, J.L.; Lodish, H. Enhanced Phosphocholine Metabolism Is Essential for Terminal Erythropoiesis. Blood 2018, 131, 2955–2966. [Google Scholar] [CrossRef]
- Pujalte, G.G.A.; Maynard, J.R. The Increasing Importance of Sports Science and Medicine. J. Int. Med. Res. 2020, 48, 030006051982769. [Google Scholar] [CrossRef]
- Tanisawa, K.; Wang, G.; Seto, J.; Verdouka, I.; Twycross-Lewis, R.; Karanikolou, A.; Tanaka, M.; Borjesson, M.; Di Luigi, L.; Dohi, M.; et al. Sport and Exercise Genomics: The FIMS 2019 Consensus Statement Update. Br. J. Sports Med. 2020, 54, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Durussel, J.; Shurlock, J.; Mooses, M.; Fuku, N.; Bruinvels, G.; Pedlar, C.; Burden, R.; Murray, A.; Yee, B.; et al. Validation of Whole-Blood Transcriptome Signature during Microdose Recombinant Human Erythropoietin (RHuEpo) Administration. BMC Genom. 2017, 18, 817. [Google Scholar] [CrossRef]
- Gupta, N.; Zhao, Y.-Y.; Evans, C.E. The Stimulation of Thrombosis by Hypoxia. Thromb. Res. 2019, 181, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Sallis, R.; Young, D.R.; Tartof, S.Y.; Sallis, J.F.; Sall, J.; Li, Q.; Smith, G.N.; Cohen, D.A. Physical Inactivity Is Associated with a Higher Risk for Severe COVID-19 Outcomes: A Study in 48 440 Adult Patients. Br. J. Sports Med. 2021, 55, 1099–1105. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glotov, A.S.; Zelenkova, I.E.; Vashukova, E.S.; Shuvalova, A.R.; Zolotareva, A.D.; Polev, D.E.; Barbitoff, Y.A.; Glotov, O.S.; Sarana, A.M.; Shcherbak, S.G.; et al. RNA Sequencing of Whole Blood Defines the Signature of High Intensity Exercise at Altitude in Elite Speed Skaters. Genes 2022, 13, 574. https://doi.org/10.3390/genes13040574
Glotov AS, Zelenkova IE, Vashukova ES, Shuvalova AR, Zolotareva AD, Polev DE, Barbitoff YA, Glotov OS, Sarana AM, Shcherbak SG, et al. RNA Sequencing of Whole Blood Defines the Signature of High Intensity Exercise at Altitude in Elite Speed Skaters. Genes. 2022; 13(4):574. https://doi.org/10.3390/genes13040574
Chicago/Turabian StyleGlotov, Andrey S., Irina E. Zelenkova, Elena S. Vashukova, Anna R. Shuvalova, Alexandra D. Zolotareva, Dmitrii E. Polev, Yury A. Barbitoff, Oleg S. Glotov, Andrey M. Sarana, Sergey G. Shcherbak, and et al. 2022. "RNA Sequencing of Whole Blood Defines the Signature of High Intensity Exercise at Altitude in Elite Speed Skaters" Genes 13, no. 4: 574. https://doi.org/10.3390/genes13040574
APA StyleGlotov, A. S., Zelenkova, I. E., Vashukova, E. S., Shuvalova, A. R., Zolotareva, A. D., Polev, D. E., Barbitoff, Y. A., Glotov, O. S., Sarana, A. M., Shcherbak, S. G., Rozina, M. A., Gogotova, V. L., & Predeus, A. V. (2022). RNA Sequencing of Whole Blood Defines the Signature of High Intensity Exercise at Altitude in Elite Speed Skaters. Genes, 13(4), 574. https://doi.org/10.3390/genes13040574