Abstract
Amyotrophic lateral sclerosis (ALS) is the most common type of motor neuron disease whose causes are unclear. The first ALS gene associated with the autosomal dominant form of the disease was SOD1. This gene has a high rate of rare variants, and an appropriate classification is essential for a correct ALS diagnosis. In this study, we re-evaluated the classification of all previously reported SOD1 variants (n = 202) from ALSoD, project MinE, and in-house databases by applying the ACMG-AMP criteria to ALS. New bioinformatics analysis, frequency rating, and a thorough search for functional studies were performed. We also proposed adjusting criteria strength describing how to apply them to SOD1 variants. Most of the previously reported variants have been reclassified as likely pathogenic and pathogenic based on the modified weight of the PS3 criterion, highlighting how in vivo or in vitro functional studies are determining their interpretation and classification. Furthermore, this study reveals the concordance and discordance of annotations between open databases, indicating the need for expert review to adapt the study of variants to a specific disease. Indeed, in complex diseases, such as ALS, the oligogenic inheritance, the presence of genes that act as risk factors and the reduced penetration must be considered. Overall, the diagnosis of ALS remains clinical, and improving variant classification could support genetic data as diagnostic criteria.
1. Introduction
Neurodegenerative diseases are a heterogeneous set of diseases of the central nervous system, characterized by the progressive degeneration of neurons’ structure and function. ALS, also known as Charcot disease or Lou Gehrig’s disease, is the most common motor neuron disease with a global annual incidence ranging from 1 to 3 cases per 100,000 population [1]. It is characterized by the progressive neuromuscular junctions dismantling and the motor neurons degeneration in the motor cortex, brain stem, and spinal cord. Loss of motor neurons leads to progressive paralysis and death from respiratory failure within 3–5 years of the onset of disease [2]. To date, despite ALS being known to be a multifactorial disease associated with genetic predisposition, environmental factors, and lifestyle, its causes are unclear. In 10% of cases, ALS is caused by a genetic mutation that can characterize several generations of the same family (familial ALS (fALS)) or the disease arises suddenly and without any known familial link (sporadic ALS (sALS)) [3]. The main genes directly associated with the onset of the disease and which account for almost 50% of fALS and 6% of sALS cases in the world are SOD1, TARDBP, FUS, and C9orf72 [4].
The first ALS gene associated with the adult-onset autosomal dominant form of the disease was mapped to the chromosome 21q22.1–22.2 and encodes for the cytoplasmic Cu/Zn superoxide dismutase (SOD1). Mutations in this gene were first described and linked to the disease in 1993 [5], and their frequencies range from 1% to 2% and from 13% to 20% in sALS and fALS patients, respectively [6]. SOD1 gene encodes a homodimer enzyme composed of 154 highly conserved amino acids between species. SOD1 mutants show a gain of function in which the protein appears to take on a new toxic function, probably related to an increased tendency for mutated proteins to assemble and form aggregates in motor neurons [7].
The advancement of next-generation sequencing (NGS) technologies and the extension of screening tests have made it possible to associate over 100 genes to the disease and thousands of variants [8]. The enormous increase in data and the massive number of unknown variants obtained using these approaches require an interpretative step that allows for clinical classification of the variants as pathogenic or benign. The guidelines proposed in 2015 by the American College of Medical Genetics and Genomics and by the Association for Molecular Pathology (ACMG-AMP) [9] represent the criteria to be used for the classification of variants associated with Mendelian disorder in different categories—namely, benign, likely benign, variants of uncertain significance, likely pathogenic, and pathogenic. Among all, one of the criteria considered very important is based on the characterization of gene variants through functional studies (PS3), and the studies in which experimental tests are carried out are fundamental, as they help the interpretation and classification of pathogenic gene variants.
Since 2015, other publications have highlighted the need to re-evaluate some criteria in the classification of variants [10,11,12]. In particular, detailed guidance was provided to determine the appropriate strength of evidence of functional studies that can be used in the interpretation of clinical variants [13]. However, despite the introduction of these dedicated guidelines and algorithms, the variant interpretation process is complex and not always an efficient process. In addition, the existence of discordant variant classifications across public databases is one of the well-documented limitations in the interpretation of the pathogenicity of variants.
Based on this premise, a periodic re-evaluation of the genetic variants underlying complex diseases is necessary, especially in complex diseases such as ALS where mutation–phenotype correlations are growing rapidly.
To date, over 200 SOD1 variants have been identified. Most are present in heterozygosity [14], confirming the autosomal dominant inheritance of the gene in this motor neuron disease [15], while others represent an exception, such as the recessive variant D91A (p.Asp91Ala) that shows frequent homozygosity in Scandinavian populations and Nordic countries [16] and the compound heterozygous variant D97N (p.Asp97Asn) [17].
This study provides an updated evaluation of all previously reported SOD1 variants in the Amyotrophic Lateral Sclerosis Online Genetics Database (ALSoD, https://alsod.ac.uk/, 20 December 2021), project MinE (http://databrowser.projectmine.com/, 22 December 2021) and in-house databases [18] by applying the ACMG-AMP guidelines with criteria adapted for SOD1. We also assess the concordance of annotations between open databases (ClinVar and LOVD), highlighting ongoing sources of discordance.
2. Materials and Methods
2.1. Structured Search
SOD1 variants associated with ALS were extrapolated from the ALSoD, project MinE, and in-house databases [18]. All these substitutions were from patients diagnosed with definite, probable, and probable lab-supported ALS based on the revised El Escorial criteria [19]. We investigated all the genetic variants listed by ALSoD, while we selected only the variants matching the “LoF + Missense” criterion listed by Project MinE. In addition, a PubMed literature search was conducted using the keywords “SOD1 variant” and “SOD1 mutation”. Studies aimed at investigating the functional significance of genetic variants and abstracts related to genetic variants underlying ALS were also considered in the study.
2.2. ACMG-AMP Pathogenicity Analysis
The analysis and clinical classification of ALS variants were based on the ACMG-AMP guidelines (Supplementary Table S1). The assigned criteria were then combined, according to the scoring rules expressed in Supplementary Figure S1, to choose a classification from the 5-level system.
2.3. Frequency Analysis of Variants
For the evaluation of the minor allelic frequency (MAF), gnomAD v2.1 (gnomad.broadinstitute.org) was used. According to the ACMG-AMP guidelines, if a variant exists in a large general population or a control cohort, it can be considered a criterion 2 moderate pathogen (PM2). Recently, Oza et al. [20] recommended downgrading the weight of the PM2 criterion to a supporting weight in guidance from the Sequence Variant Interpretation (SVI) working group (https://clinicalgenome.org/site/assets/files/5182/pm2_-_svi_recommendation_-_approved_sept2020.pdf, 27 January 2022). In our context, SOD1 variants completely absent from all databases or with a MAF < 0.001 met the moderate pathogen criterion 2 (PM2) [21,22].
2.4. Computational Tools Analysis
The pathogenicity of the variants was also evaluated using multiple in silico genomic tools, including SIFT [23], Polyphen-2 [24], MutationTaster [25], MutationAssessor [26], FATHMM [27], PROVEAN [28], and CADD [29]. Splicing prediction analysis was performed using SpiP and SpliceAI tools [30]. The convergence should be considered as supporting evidence of pathogenicity or benignity. An adverse consequence predicted with more than six tools is considered to be evidence to support pathogenicity (PP3), while at least three total and concordant predictors are required to support a benign state (BP4). However, if the in silico predictions disagree, this test should not be used for the classification of a variant.
2.5. Analysis of Functional Studies
Functional studies conducted in vitro or in vivo may show a detrimental effect on protein function, supporting the pathogenicity of the tested variant (PS3), or no effect, thus supporting a benign impact of the variant (BS3). As recommended by Brnich et al., several factors must be considered for the assignment of this criterion [13]. It is necessary to delineate the genetic mechanism of the disease to choose the appropriate functional analysis, as well as the experimental model to be used on which to perform functional tests. Assays performed must be coded in biological replicates, and an adequate number of control cases should be considered. Finally, the test result must be in line with the pathogenetic mechanism of the disease [13].
ALS is a complex biological system, and the precise mechanisms underlying selective cell death in the disease are unknown. Current understanding indicates that there may be a complex interplay between genetic factors, oxidative stress, excitotoxicity, protein aggregation, impaired axonal transport, and mitochondrial damage. Over the years, several in vitro and in vivo experimental models have been exploited to study the molecular processes underlying motor neuron degradation and disease progression, providing essential small pieces to the ALS puzzle [31,32]. However, only an integrated and complimentary use of different experimental models could improve our understanding of the pathogenic mechanism of the disease.
According to Brnich’s recommendations and experimental evidence on SOD1 variants, we assigned three different weights to the PS3 criterion: PS3_Strong, PS3_Moderate, and PS3_Supporting. PS3_Strong was assigned to variants in which in vivo functional assay results showed toxic SOD1 function and disruption of mitochondrial dynamics, protein folding, axonal transport, and cellular metabolism. The PS3_Moderate and PS3_Supporting criteria were assigned to variants investigated by functional in vitro studies, following the flowchart shown in Supplementary Figure S2.
2.6. Critical Functional Domain Position
Pathogenic SOD1 variants associated with ALS are spread over the entire length of the gene. Hotspot mutations are characterized by the presence of many missense variants, in-frame, and nonsynonymous variants, most of which have a pathogenicity classification. The universal protein source, UniProt (https://www.uniprot.org/, 29 January 2022), and bioinformatics tools can help identify mutations in established hotspots or functional domains. The PM1 rule considers reported variants within the domain and is assigned with the ratio pathogenic to total nonpathogenic variants is greater than 1.5, with at least 5 pathogenic variants within the domain. We established that the PM1 criterion can be upgraded to strong (PM1_Strong) when at least 14 pathogenic variants are reported in the functional domain; the moderate weight (PM1_Moderate) of this criterion was assigned in the presence of at least 7 pathogenic missense variants. Finally, we considered PM1_Supporting when 5–6 pathogenic mutations are reported in the domain (Supplementary Table S2). The distribution of SOD1 variants is shown in Figure 1A.
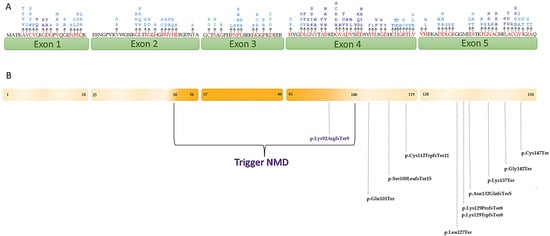
Figure 1.
(A) Schematic representation of SOD1 showing the localization of variants that were assigned PM1. The amino acid changes are represented in red. The different weights of the criterion are represented by the different colors: dark blue, PM1_Strong; light blue, PM1_Moderate; green, PM1_Supporting. The numbers under the exons represent the amino acid distribution; (B) localization of variants that undergo/do not undergo NMD. The variants localized in SOD1 region where PTCs trigger NMD are represented in purple, the variants localized in the region that do not undergo NMD are colored in black.
2.7. Evaluation of the PVS1 Criterion
A strong weight in the classification of ALS genes is represented by the PVS1 criterion. This is attributed to a “Null variant in a gene where loss of function (LOF) is a known mechanism of disease”. Gain of function and several mechanisms are confirmed to be involved in ALS [33,34,35]. However, much evidence about SOD1 loss of function (LOF) suggests it may play a role in ALS pathogenesis, probably increasing the susceptibility to neurodegeneration. Overall, the major effect of SOD1 mutations in ALS is linked to the protein aggregation but these may also lead to loss of function of SOD1 activity or loss of the nuclear function, where SOD1 acts as a transcription factor [6].
According to the recommendations provided by the ClinGen SVI working group [10] and taking into account Guissart’s considerations [36], we adapted ACMG-AMP guidelines for SOD1 by evaluating the right strength related to the PVS1 criterion and distinguishing the variants with PVS1_Very strong and PVS1_Strong. In particular, we assigned PVS1_Very strong criterion when variants do not undergo the nonsense-mediated mRNA decay (NMD) and PVS1_Strong when variants undergo NMD. A representation of the SOD1 gene showing the localization of premature termination codons (PTCs) that cause ALS and NMD escaping is reported in Figure 1B.
2.8. Comparison between Databases
To assess the concordance between annotations generated by ClinVar and LOVD databases, we compared a list of 202 variants of the SOD1 gene across several online resources. The databases investigated generate an automated interpretation of the variants. Specifically, Leiden Open Variation Database (LOVD—https://www.lovd.nl/, 5 January 2022) is a software database for collecting and visualizing variants in the DNA sequence [37]. ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/, 7 January 2022) is an archive that aggregates information on genomic variation and the various correlations with human health [38].
2.9. Statistical Analysis
The chi-square concordance test was performed for discordance–concordance and variant classification across the various databases, using the SPSS 20.0 test (SPSS, Chicago, IL, USA). Pathogenic/likely pathogenic SOD1 variants were compared using Fisher’s test. A p-value < 0.05 was considered statistically significant.
3. Results
3.1. Summary of Variants
In this study, 202 sequence variants of SOD1 (NM_000454.5) were collected and analyzed. These variants have a MAF < 0.001 considering gnomAD 2.1 populations. Table 1 lists all variant sequences investigated and corresponding classification according to the ACMG-AMP guidelines.

Table 1.
SOD1 variants in Amyotrophic Lateral Sclerosis: Systematic re-evaluation according to ACMG-AMP guidelines.
The variants analyzed were five splice regions (2%), one nonsense (1%), eight frameshifts (4%), three stop-gained (1%), one synonymous (1%), and three intron variants (1%), while all remaining were missense variants (90%) (Figure 2).
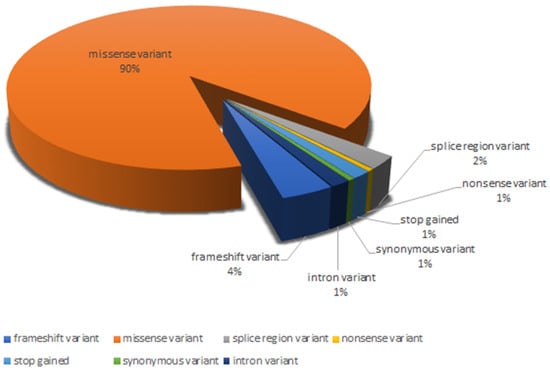
Figure 2.
Distribution of the different variant types analyzed.
In total, 13 variants (6.4%) were classified as variants of uncertain significance (VUS), with insufficient or conflicting evidence regarding the role of molecular alteration in disease. Of these, nine were missense variants (c.59A>G, c.89T>C, c.179G>A, c.298A>G, c.172T>C, c.179G>T, c.328G>T, c.35A>C, c.172T>G), three were intron variants (c.358-11A>G, c.358-304C>G, c.358-10T>G) and one was a splice region variant (c.240-7T>G). The variants classified as likely pathogenic were 136 (67.3%), of which 2 were frameshifts (1.5%), 1 was stop-gained (0.7%), 3 were splice region variants (2.2%), and 130 were missense variants (95.6%). The remaining 53 (26.3%) were considered pathogenic and included 6 frameshifts (11.3%), 1 splice region (1.9%), 2 nonsense (3.8%), 2 stop-gained (3.8%), 1 synonymous variant (1.9%), and 41 missense variants (77.3%).
3.2. Contribution of Each Criterion to the Final Classification
The contribution of each criterion to the final classification is shown in Figure 3. The PM2 criterion was assigned to all 202 variants analyzed in this study. Pathogenic variants of the SOD1 gene are often identified in a single patient or an isolated family. This evidence underlines the rarity of the variants and therefore allows the PM2 criterion to acquire a moderate level of evidence of pathogenicity.
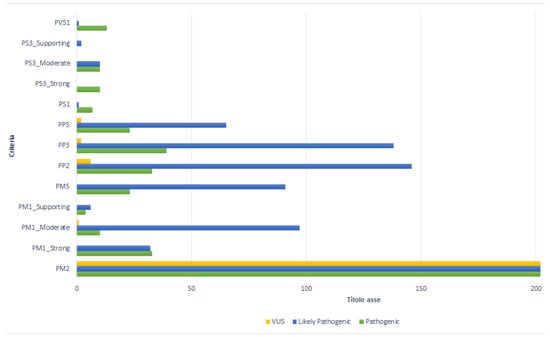
Figure 3.
Contribution of each criterion to the final classification.
The PM1 criterion was assigned to 186 variants (92%), with different weights depending on the number of pathogenic variants located in the functional domain of the gene. The PM1_Strong rule was assigned to 68 variants (36.6%), of which 32 (47.1%) were classified as likely pathogenic, 33 (48.5%) as pathogenic, and 3 (4.4%) as VUS. The PM1_ Moderate was assigned to 108 variants (58%); in this category, 97 (89.9%) were likely pathogenic, 10 (9.3%) as pathogenic, and only 1 (0.9%) was classified as VUS. The PM1_Supporting weight was assigned to 10 mutations (5.4%), of which 6 terms (60%) were classified as likely pathogenic and 4 (40%) as VUS.
The PP2 criterion characterized 185 variants (91.6%): 146 (79%) as likely pathogenic, 33 (17.9%) as pathogenic, and 6 (3.1%) as VUS.
Computational tools predicted a disease-causing role (PP3) in 179 (88.6%) cases examined. Of these variants, 138 variants (77%) were classified as likely pathogenic, 39 (21.8%) as pathogenic, and 2 (1.1) as VUS.
In the present study, the PM5 criterion assigned to a “novel missense change at an aminoacidic residue where a different missense change determined to be pathogenic has been seen before” [9], showed a moderate impact in 114 variants (56.4%). Of these, 91 (79.8%) were likely pathogenic, while the remaining 23 (20%) were pathogenic.
According to the ACMG-AMP guidelines, the PP5 criterion was assigned to 90 variants (90/202; 44.6%) for which different studies were available in the literature. The presence of the PP5 criterion allowed the classification of 65 (72.2%) likely pathogenic, 23 (25.5%) pathogenic, and of the remaining 2 (2.2%) VUS variants. The splice region c.240-7T>G and the intron c.358-11A>G variants were characterized as VUS, with the PM2 and PP5 criteria, since no data from the literature were available.
Considering previously published functional studies for the evaluation of the variant effect and the impact of the mutation on the clinical phenotype, the PS3 criterion was assigned to 32 (32/202; 15.8%) variants (Supplementary Table S3). PS3_Strong was assigned to 10 (31.3%) variants classified as pathogenic. These pathogenic variants were characterized by the attendance of multiple functional studies performed in various experimental models (Supplementary Table S3) [39,40,41,42,43,44,45,46,47,48,49]. The p.Gly94Ala mutation, known as G93A, was used to create the first ALS-associated SOD1 transgenic mouse. This model, which expresses large amounts of mutant SOD1, develops adult-onset neurodegeneration of spinal motor neurons and progressive motor deficits leading to paralysis, and it is now widely used both for pathogenic molecular mechanisms and for preclinical studies useful for the specific therapy of affected patients [31,39,40,41]. This mutation is currently the one studied in the largest number of animal models including C. Elegans, Saccharomyces Cerevisiae, Swine, and Danio rerio (Zebrafish) models. In the same way, experimental models of the transgenic mouse G93A and H46R (p.His47arg) show degeneration of the upper and lower motoneurons associated with a gravity directly proportional to the altered protein expression [31,39,42]. In addition, in vivo experiments show that SOD1-D91A (p.Asp91Ala), the most common mutation affecting SOD1 linked to ALS, is less toxic than other tested mutants, while homozygous mice develop fatal motor neuron disease similar to that observed in homozygous-D91A human ALS patients [41,43].
A Zebrafish model with G94R (p.Gly94Arg) variant has been developed, and this shows a mutated expression of SOD1 at a physiological level, which allows targeted pre-clinical studies [31,49]. For the variants G86R (p.Gly86Arg) [31,45,46,47] and G38R (p.Gly38Arg) [31,47,48], there are several functional studies carried out on transgenic mouse models in which there are high expression levels of the mutated gene and differences in the age of onset and progression of the disease as well as lifespan. For the G86R variant, studies have also been carried out using the C. Elegans nematode model. In particular, the presence of this missense variant produces an abnormal function of neurons ad a cellular dysfunction at the muscular level. The same variant was tested on Drosophila melanogaster models showing a progressive deterioration of motor functions with a specific clinical phenotype. The same experimental model was also used to study the effect of variants G38R and I114T (p.Ile114thr), determining a decrease in the lifespan of the midge accompanied by abnormalities in movement (Supplementary Table S3).
The effect of other mutations, classified with the PS3_Moderate or PS3_Supporting criterion, was evaluated by functional in vitro studies [50,51,52,53,54,55,56,57,58,59,60,61,62,63] assessing the correlation between the specific genetic alteration and the modification of oxidative stress, mitochondrial and cellular metabolism in different clinical phenotypes (Supplementary Table S3). PS3 with moderate weight was attributed to 20 (62.5%) variants: 10 (50%) were classified as likely pathogenic, while the remaining 10 (50%) as pathogenic. Finally, the PS3_Supporting weight was assigned to two (6.2%) variants classified as likely pathogenic (Supplementary Figure S2).
Another criterion with a strong impact on the classification, the PS1, was assigned to only eight SOD1 variants (4%). Of these, seven (87.5%) were classified as pathogenic and one (12.5%), the c.355G>T, was classified as likely pathogenic.
In our study, it was possible to attribute the PVS1 rule to 11 (5.4%) variants, 10 classified as pathogenic (92.8%) and 1 as likely pathogenic (7.1%). Only the variant c.275_276delAA expected to undergo NMD, while the other 10 mutations (c.301G>T, c.441T>A, c.409A>T, c.380T>A, c.424G>T, c.383_384insACCC, c.384_385insTGGG, c.320_321insT, c.335dup, c.383_392dup) did not undergo NMD, presenting the PVS1_Very strong criterion. The stop-gain variant c.301G>T, although known as a pathogenic mechanism (PVS1) and absent from population databases (PM2), was classified as likely pathogenic (Supplementary Table S4).
The frameshift variants c.272_274dupACA and c.56_58delTCA cause a change in the length of the protein for which the PM4 criterion can be applied. Combined with the PM and PP criteria, these two variants were classified as likely pathogenic.
3.3. Concordance–Discordance between Open Databases and Reclassification Based on ACMG-AMP Guidelines
LOVD contains a total of 47 variants, of which only 18 are classified as pathogenic, likely pathogenic, or VUS. The comparison of these variants (18/202, 9%) revealed a discordant classification (p-value < 0.001) in two substitutions. The variant c.59A>G, defined as likely benign in LOVD databases, was reclassified as VUS, while the c.272A>C was reclassified from VUS to pathogenic (Supplementary Table S5).
Evaluating SOD1 variant classifications in ClinVar database, 72 (35.6%) were concordant, and 19 (9.4%) variants were discordant (p-value < 0.001). In particular, 12 VUS were reclassified as likely pathogenic (n = 10) and pathogenic (n = 3), while 1 pathogenic variant was reclassified as VUS. Seven variants, defined as conflicting interpretations of pathogenicity by ClinVar, were reclassified: six as likely pathogenic/pathogenic and only one as VUS (Supplementary Table S5). Finally, 111 terms (55%), searched for rs ID, did not match in ClinVar.
4. Discussion
In this study, for the first time, ALS-associated SOD1 variants were systematically re-evaluated to perform clinical interpretation according to the ACMG-AMP guidelines. We also provided an updated assessment of variant concordance–discordance between bioinformatics tools used to evaluate variants’ roles and the criteria application provided by the above-mentioned guidelines adapted for SOD1.
Previous research has reported that only 34% of variants have a concordant classification according to the ACMG-AMP guidelines [64]. The high percentage of discordance between classifications can be explained by how individual laboratories interpret and analyze variants data, based on published levels of evidence and the misuse of ACMG-AMP guidelines.
Various scientific evidence supports the idea that ALS is caused by a combination of genetic and environmental factors [65,66]. To understand and quantify the value of the contribution of genetics in the pathogenesis of the disease several studies have been conducted [46]. Although a large number of genes have been related to ALS, other variants are to be discovered, which could play adjunct roles in increasing risk or susceptibility to the disease. Specific mutations not identifiable by GWAS or massive sequencing methods, such as repeated expansions [67] or variants in noncoding regions, could belong to this category [68,69].
Human SOD1 is widely known to be an important protein in the antioxidant system. The main hallmark of fALS is the presence of SOD1 aggregates in the spinal cord of patients [70], and the chemically modified form of the protein in the spinal cord of sporadic ALS patients has also been found [71]. SOD1 mutations were the first to be directly associated with the disease pathogenesis and the classification of variants in such a complex pathology remains a challenge. The heterogeneity of clinical data and the difference in approaches used for the interpretation of variants are the main challenges in variant classification [72].
SOD1 gene has a high rate of rare variants, and an appropriate classification is essential for a correct ALS diagnosis. Furthermore, one of the greatest challenges in interpreting pre-existing assessments is the heterogeneity in the approaches used for variant analysis. In this study, we applied a standardized in silico analysis and assessed the frequency of the variants, as well as considering the presence of functional and segregation studies. Results showed that 136 (67.3%) variants met the criteria of likely pathogenic, 53 (26.3%) were classified as pathogenic (Figure 4), and the remaining variants (n = 13; 6.4%) were categorized as VUS.
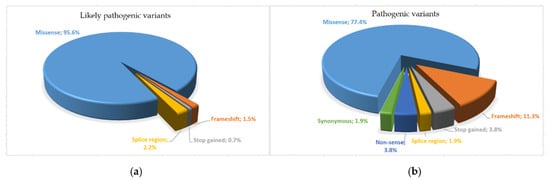
Figure 4.
Pie chart of different variant distribution types identified: (a) likely pathogenic variants; (b) pathogenic variants.
In our study, the PS3 criterion proved to be fundamental for the pathogenicity classification of 32 variants, representing 62.5% of total pathogenic terms. However, the near absence of functional and segregation data for a large number of terms remains an important limiting factor, considering that functional evidence is the main scoring driver for pathogenic/likely pathogenic for missense variants. Compared with all the variants analyzed, it is clear that there are few functional studies, conducted in vitro or in vivo, which show a detrimental effect on the protein. Furthermore, the PS3 criterion is not automatically assigned in the various databases, as functional evidence must be searched by the geneticist consulting PubMed and other available sites (Mastermind, etc.). After the manual revision, the strength of the criterion can also be changed according to the relevance of the evidence.
The attribution of the PM1 criterion was based on scientific evidence showing the absence of specific mutational hotspot mutations in SOD1 linked to ALS, indicating the whole protein involved [73]. In SOD1, several variants have been described to cluster in all five exons of the gene identified as hotspots (Figure 1A). Rather than defining clear function domains, these regions are defined by an increase in pathogenic variations identified in ALS patients. For this reason, we considered three different levels of strength of the criterion: PM1_Strong (≥14 variants), PM1_Moderate (at least 7 variants), and PM1_Supporting (5–6 variants).
All of the variants present in this study with a MAF value less than 0.001 and absents from all databases were considered to have a moderate pathogenicity level. The definition of a MAF threshold for ALS, in which significant confounding factors coexist (late-onset of the disease, age-dependent, variant-related reduced penetrance, and oligogenic inheritance), cannot ignore many factors difficult to evaluate, such as the inheritance model (monoallelic for most SOD1-ALS variants), the prevalence of the disease, and the penetrance. Failing consensus on allele frequency thresholds, the PM2 criterion was assigned to these cases.
For most of the missense variants involved in our study and classified as pathogenic or likely pathogenic, the PP5 criterion was considered supporting evidence with a very strong value, as the variants are present in several studies in the literature. However, this criterion must be used with caution as “the evidence is not available to the laboratory to perform an independent evaluation“ [11].
A strong impact on the evaluation of a mutation is expressed by the PS1 criterion, which is attributed when the same amino acid variation of a well-known causative variant occurs. The variant could act directly through the specific change of DNA or could modify the protein sequence through various mechanisms, such as an alteration in splicing. To support the pathogenicity of genetic variants, it is necessary to have well-established and reproducible functional studies.
The PVS1 criterion is considered the greatest impact, characterized by identifying a variant that causes a loss-of-function (LoF) allele. Although the gain of function is causative for amyotrophic lateral sclerosis, SOD1 loss of function may play a role in ALS, contributing to the onset and progression of the disease [73,74]. Many nonsense or frameshift mutations may lead to a LOF of SOD1 by affecting its structures and/or interactions pattern [6]. mRNA containing premature termination codons are known to be rapidly degraded by nonsense-mediated mRNA decay and premature termination codons that escape NMD in SOD1, causing the production of truncated SOD1 protein, which may have high toxicity [36] (Figure 1B).
An element of increasing importance is constituted by databases that report identified variants with their associated phenotypes and the interpretation of their functional role. In ALS, this concept is even more important since the vast majority of the variants identified are new and/or private, and for this reason, it is very important to share the knowledge obtained in dedicated databases. Another aspect to note is the presence/absence of SOD1 variants in these different databases. This study shows that many variants of this gene are absent in online archives used and, therefore, stresses that it is necessary to revise these data, so they are also open source, allowing the scientific community to search for updated information of genetic mutations, which are extremely important to the disease. A comparison of the annotations between open databases and our reclassification based on ACMG-AMP guidelines with the criteria adapted for SOD1 revealed a different rate of discordance. In particular, comparing ClinVar, we found discordance between pathogenic/likely pathogenic variants and VUS in 9.4% of terms. A case in point is the most common SOD1 mutation, p.Asp91Ala, which is still reported in ClinVar with conflicting interpretations of pathogenicity despite extensive evidence demonstrating pathogenicity according to the ACMG-AMP guidelines. In this public repository, many variants are associated with classification according to the ACMG-AMP guidelines. This archive accepts data derived from different working groups, the relevant literature, and documents from other web resources. To ensure the quality of the declarations, a review phase is carried out that involves evaluation by groups of experts. The variant data are then updated based on the new information presented, and therefore, an adequate time is required for the process. [39].
Based on the data obtained, it is evident that the mere application of the guidelines for interpreting genetic variants developed and refined so far cannot be considered sufficient in ALS to address various problems related to the implementation of new genetic analysis technologies in a diagnostic and clinical context. A manual reinterpretation of variants is essential to adapt the study to a particular disease, considering the specific characteristics of the gene and allowing for a consistent application. In this way, the different ACMG-AMP criteria acquire a different weight based on the type of pathology, scientific evidence, and knowledge of the variant itself.
5. Conclusions
In this study, we re-evaluated the classification of all previously reported SOD1 variants (n = 202) in ALSoD, Project MinE, and in-house databases by applying the ACMG-AMP criteria to ALS. Our results underline the importance of classifying variants involved in ALS and SOD1 variants with caution.
Specific guidelines for ALS would allow a more adequate classification of the large number of variants of uncertain significance, which have been constantly discovered. This can be accomplished by carrying out genetic research on large cohorts of patients and controls and by sharing the data obtained with the scientific community. Furthermore, it is crucial to develop and implement new analysis strategies that could facilitate the discovery and the validation of genetic factors involved in the pathogenesis of this disease that is still unknown. To date, the diagnosis of ALS remains, based on clinical, not genetic criteria. The improvement of the existing variant classification systems available could provide a better basis for using genetic findings as diagnostic criteria.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/genes13030537/s1. Table S1: The ACMG-AMP criteria and their relevance in ALS, Table S2: Assessment of the PM1 criterion in SOD1 variants, Table S3: List of in vivo and in vitro functional studies considered for the PS3 criterion attribution, Table S4: Characteristics of the SOD1 variants relevant to the PVS1 criterion, Table S5: List of SOD1 variants and concordance–discordance between open databases and reclassification, Figure S1: ACMG-AMP guidelines for combining criteria to classify sequencing variants, Figure S2: Decision tree for functional evidence PS3.
Author Contributions
Conceptualization, P.R. and F.L.C.; methodology, P.R.; formal analysis, P.R.; resources, P.R. and B.P.; data curation, P.R.; writing—original draft preparation, P.R.; writing—review and editing, P.R. and F.L.C.; supervision, F.L.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a special award to the Department of Pharmacy, Health and Nutritional Sciences of University of Calabria (Italy) (Department of Excellence, Italian Law 232/2016) from the Italian Ministry of Education, University and Research (MIUR) and by MIUR ex 60% to F.L.C.
Institutional Review Board Statement
This study did not require ethical approval.
Informed Consent Statement
This study did not involve humans.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to thank the Project MinE GWAS Consortium.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Riancho, J.; Delgado-Alvarado, M.; Andreu, M.D.; Paz-Fajardo, L.; Arozamena, S.; Gil-Bea, F.J.; de Munaín, A.L. Amyotrophic lateral sclerosis (ALS), cancer, autoimmunity and metabolic disorders: An unsolved tantalizing challenge. Br. J. Pharmacol. 2021, 178, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Liu, T.; Liu, L.; Yao, X.; Chen, L.; Fan, D.; Zhan, S.; Wang, S. Global variation in prevalence and incidence of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. 2020, 267, 944–953. [Google Scholar] [CrossRef] [PubMed]
- Ruffo, P.; Strafella, C.; Cascella, R.; Caputo, V.; Conforti, F.L.; Andò, S.; Giardina, E. Deregulation of ncRNA in Neurodegenerative Disease: Focus on circRNA, lncRNA and miRNA in Amyotrophic Lateral Sclerosis. Front. Genet. 2021, 12, 784996. [Google Scholar] [CrossRef] [PubMed]
- Perrone, B.; Conforti, F.L. Common mutations of interest in the diagnosis of amyotrophic lateral sclerosis: How common are common mutations in ALS genes? Expert Rev. Mol. Diagn. 2020, 20, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Berdyński, M.; Miszta, P.; Safranow, K.; Andersen, P.M.; Morita, M.; Filipek, S.; Żekanowski, C.; Kuźma-Kozakiewicz, M. SOD1 mutations associated with amyotrophic lateral sclerosis analysis of variant severity. Sci. Rep. 2022, 12, 1–11. [Google Scholar] [CrossRef]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef]
- Lattante, S.; Marangi, G.; Doronzio, P.N.; Conte, A.; Bisogni, G.; Zollino, M.; Sabatelli, M. High-Throughput Genetic Testing in ALS: The Challenging Path of Variant Classification Considering the ACMG Guidelines. Genes 2020, 11, 1123. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M.; ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef]
- Biesecker, L.G.; Harrison, S.M.; e ClinGen Sequence Variant Interpretation Working Group. The ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genet. Med. 2018, 20, 1687–1688. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Harrison, S.M.; Rehm, H.L.; Plon, S.E.; Biesecker, L.G.; on behalf of ClinGen Sequence Variant Interpretation Working Group. Updated recommendation for the benign stand-alone ACMG/AMP criterion. Hum. Mutat. 2018, 39, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Brnich, S.E.; On behalf of the Clinical Genome Resource Sequence Variant Interpretation Working Group; Tayoun, A.N.A.; Couch, F.J.; Cutting, G.R.; Greenblatt, M.S.; Heinen, C.D.; Kanavy, D.M.; Luo, X.; McNulty, S.M.; et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 2019, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.M.; Nilsson, P.; Forsgren, L.; Marklund, S.L. CuZn-Superoxide Dismutase, Extracellular Superoxide Dismutase, and Glutathione Peroxidase in Blood from Individuals Homozygous for Asp90Ala CuZn-Superoxide Dismutase Mutation. J. Neurochem. 1998, 70, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Muratet, F.; Teyssou, E.; Chiot, A.; Boillée, S.; Lobsiger, C.S.; Bohl, D.; Gyorgy, B.; Guegan, J.; Marie, Y.; Amador, M.D.M.; et al. Impact of a frequent nearsplice SOD1 variant in amyotrophic lateral sclerosis: Optimising SOD1 genetic screening for gene therapy opportunities. J. Neurol. Neurosurg. Psychiatry 2021, 92, 942–949. [Google Scholar] [CrossRef]
- Gentile, G.; Perrone, B.; Morello, G.; Simone, I.L.; Andò, S.; Cavallaro, S.; Conforti, F.L. Individual Oligogenic Background in p.D91A-SOD1 Amyotrophic Lateral Sclerosis Patients. Genes 2021, 12, 1843. [Google Scholar] [CrossRef]
- Hand, C.K.; Mayeux-Portas, V.; Khoris, J.; Briolotti, V.; Clavelou, P.; Camu, W.; Rouleau, G.A. Compound Heterozygous D90A and D96N SOD1 Mutations in a Recessive Amyotrophic Lateral Sclerosis Family. Ann. Neurol. 2001, 49, 267–271. [Google Scholar] [CrossRef]
- Ungaro, C.; Sprovieri, T.; Morello, G.; Perrone, B.; Spampinato, A.G.; Simone, I.L.; Trojsi, F.; Monsurrò, M.R.; Spataro, R.; La Bella, V.; et al. Genetic investigation of amyotrophic lateral sclerosis patients in south Italy: A two-decade analysis. Neurobiol. Aging 2021, 99, 99.e7–99.e14. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L.; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A.; et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef]
- Li, J.; Zhao, T.; Zhang, Y.; Zhang, K.; Shi, L.; Chen, Y.; Wang, X.; Sun, Z. Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Res. 2018, 46, 7793–7804. [Google Scholar] [CrossRef] [PubMed]
- Davieson, C.D.; Joyce, K.E.; Sharma, L.; Shovlin, C.L. DNA variant classification–reconsidering “allele rarity” and “phenotype” criteria in ACMG/AMP guidelines. Eur. J. Med. Genet. 2021, 64, 104312. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Day, I.N.M.; Gaunt, T.R. Predicting the functional consequences of cancer-associated amino acid substitutions. Bioinformatics 2013, 29, 1504–1510. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Baux, D.; Van Goethem, C.; Ardouin, O.; Guignard, T.; Bergougnoux, A.; Koenig, M.; Roux, A.-F. MobiDetails: Online DNA variants interpretation. Eur. J. Hum. Genet. 2021, 29, 356–360. [Google Scholar] [CrossRef]
- Bonifacino, T.; Zerbo, R.A.; Balbi, M.; Torazza, C.; Frumento, G.; Fedele, E.; Bonanno, G.; Milanese, M. Nearly 30 Years of Animal Models to Study Amyotrophic Lateral Sclerosis: A Historical Overview and Future Perspectives. Int. J. Mol. Sci. 2021, 22, 12236. [Google Scholar] [CrossRef] [PubMed]
- Myszczynska, M.; Ferraiuolo, L. NewIn VitroModels to Study Amyotrophic Lateral Sclerosis. Brain Pathol. 2016, 26, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.J.; Talbot, K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog. Neurobiol. 2008, 85, 94–134. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non–cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann. Neurol. 2009, 65, S3–S9. [Google Scholar] [CrossRef]
- Guissart, C.; Mouzat, K.; Kantar, J.; Louveau, B.; Vilquin, P.; Polge, A.; Raoul, C.; Lumbroso, S. Premature termination codons in SOD1 causing Amyotrophic Lateral Sclerosis are predicted to escape the nonsense-mediated mRNA decay. Sci. Rep. 2020, 10, 1–7. [Google Scholar] [CrossRef]
- Fokkema, I.F.A.C.; Taschner, P.E.M.; Schaafsma, G.; Celli, J.; Laros, J.F.; Dunnen, J.T.D. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Nagai, M.; Aoki, M.; Miyoshi, I.; Kato, M.; Pasinelli, P.; Kasai, N.; Brown, R.H., Jr.; Itoyama, Y. Rats Expressing Human Cyto-solic Copper-Zinc Superoxide Dismutase Transgenes with Amyotrophic Lateral Sclerosis: Associated Mutations Develop Motor Neuron Disease. J. Neurosci. 2001, 21, 9246–9256. [Google Scholar] [CrossRef]
- Li, J.; Song, M.; Moh, S.; Kim, H.; Kim, D.-H. Cytoplasmic Restriction of Mutated SOD1 Impairs the DNA Repair Process in Spinal Cord Neurons. Cells 2019, 8, 1502. [Google Scholar] [CrossRef]
- Liguori, F.; Amadio, S.; Volonté, C. Where and Why Modeling Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 3977. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Nagai, M.; Aoki, M.; Komori, T.; Itoyama, Y.; Iwata, M. Motor Neuron Disease in Transgenic Mice with an H46R Mutant SOD1 Gene. J. Neuropathol. Exp. Neurol. 2007, 66, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, P.A.; Graffmo, K.S.; Brännström, T.; Nilsson, P.; Andersen, P.M.; Marklund, S.L. Motor Neuron Disease in Mice Expressing the Wild Type-Like D90A Mutant Superoxide Dismutase-1. J. Neuropathol. Exp. Neurol. 2006, 65, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-N.; Tjostheim, S.; DaSilva, K.; Taylor, D.; Zhao, B.; Rakhit, R.; Brown, M.; Chakrabartty, A.; McLaurin, J.; Robertson, J. Targeting of Monomer/Misfolded SOD1 as a Therapeutic Strategy for Amyotrophic Lateral Sclerosis. J. Neurosci. 2012, 32, 8791–8799. [Google Scholar] [CrossRef]
- Baskoylu, S.N.; Yersak, J.; O’Hern, P.; Grosser, S.; Simon, J.; Kim, S.; Schuch, K.; Dimitriadi, M.; Yanagi, K.S.; Lins, J.; et al. Single copy/knock-in models of ALS SOD1 in C. elegans suggest loss and gain of function have different contributions to cholinergic and glutamatergic neurodegeneration. PLoS Genet. 2018, 14, e1007682. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-Linked SOD1 Mutant G85R Mediates Damage to Astrocytes and Promotes Rapidly Progressive Disease with SOD1-Containing Inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef]
- Şahin, A.; Held, A.; Bredvik, K.; Major, P.; Achilli, T.-M.; Kerson, A.G.; Wharton, K.; Stilwell, G.; Reenan, R. Human SOD1 ALS Mutations in a Drosophila Knock-In Model Cause Severe Phenotypes and Reveal Dosage-Sensitive Gain- and Loss-of-Function Components. Genetics 2017, 205, 707–723. [Google Scholar] [CrossRef]
- Witan, H.; Gorlovoy, P.; Kaya, A.M.; Koziollek-Drechsler, I.; Neumann, H.; Behl, C.; Clement, A.M. Wild-type Cu/Zn superoxide dismutase (SOD1) does not facilitate, but impedes the formation of protein aggregates of amyotrophic lateral sclerosis causing mutant SOD1. Neurobiol. Dis. 2009, 36, 331–342. [Google Scholar] [CrossRef]
- Ramesh, T.; Lyon, A.N.; Pineda, R.H.; Wang, C.; Janssen, P.M.L.; Canan, B.D.; Burghes, A.; Beattie, C.E. A genetic model of amyotrophic lateral sclerosis in zebrafish displays phenotypic hallmarks of motoneuron disease. Dis. Model. Mech. 2010, 3, 652–662. [Google Scholar] [CrossRef]
- Keskin, I.; Forsgren, E.; Lange, D.J.; Weber, M.; Birve, A.; Synofzik, M.; Gilthorpe, J.D.; Andersen, P.M.; Marklund, S.L. Effects of Cellular Pathway Disturbances on Misfolded Superoxide Dismutase-1 in Fibroblasts Derived from ALS Patients. PLoS ONE 2016, 11, e0150133. [Google Scholar] [CrossRef]
- Lin, H.-X.; Tao, Q.-Q.; Wei, Q.; Chen, C.-X.; Chen, Y.-C.; Li, H.-F.; Gitler, A.D.; Wu, Z.-Y. Identification and functional analysis of novel mutations in the SOD1 gene in Chinese patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 222–228. [Google Scholar] [CrossRef]
- Prudencio, M.; Hart, P.J.; Borchelt, D.R.; Andersen, P.M. Variation in aggregation propensities among ALS-associated variants of SOD1: Correlation to human disease. Hum. Mol. Genet. 2009, 18, 3217–3226. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Amori, I.; Pesaresi, M.G.; Ferri, A.; Nencini, M.; Carrì, M.T. Cysteine 111 Affects Aggregation and Cytotoxicity of Mutant Cu,Zn-superoxide Dismutase Associated with Familial Amyotrophic Lateral Sclerosis. J. Biol. Chem. 2008, 283, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Walczak, J.; Dębska-Vielhaber, G.; Vielhaber, S.; Szymański, J.; Charzyńska, A.; Duszyński, J.; Szczepanowska, J. Distinction of sporadic and familial forms of ALS based on mitochondrial characteristics. FASEB J. 2019, 33, 4388–4403. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gutmann, D.H.; Roos, R.P. Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Hum. Mol. Genet. 2011, 20, 286–293. [Google Scholar] [CrossRef]
- Philips, T.; Rothstein, J.D. Rodent Models of Amyotrophic Lateral Sclerosis. Curr. Protoc. Pharmacol. 2015, 69, 5.67.1–5.67.21. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.P.; Rajan, S.; Duffy, L.; Mortiboys, H.; Higginbottom, A.; Grierson, A.; Shaw, P.J. Superoxide dismutase 1 mutation in a cellular model of amyotrophic lateral sclerosis shifts energy generation from oxidative phosphorylation to glycolysis. Neurobiol. Aging 2014, 35, 1499–1509. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kaneko, K.; Yamanaka, K.; O’Halloran, T.V.; Nukina, N. Complete Loss of Post-translational Modifications Triggers Fibrillar Aggregation of SOD1 in the Familial Form of Amyotrophic Lateral Sclerosis. J. Biol. Chem. 2008, 283, 24167–24176. [Google Scholar] [CrossRef]
- Salehi, M.; Nikkhah, M.; Ghasemi, A.; Arab, S.S. Mitochondrial membrane disruption by aggregation products of ALS-causing superoxide dismutase-1 mutants. Int. J. Biol. Macromol. 2015, 75, 290–297. [Google Scholar] [CrossRef]
- McAlary, L.; Yerbury, J.; Aquilina, J.A. Glutathionylation potentiates benign superoxide dismutase 1 variants to the toxic forms associated with amyotrophic lateral sclerosis. Sci. Rep. 2013, 3, 3275. [Google Scholar] [CrossRef]
- McAlary, L.; Aquilina, J.A.; Yerbury, J.J. Susceptibility of Mutant SOD1 to Form a Destabilized Monomer Predicts Cellular Aggregation and Toxicity but Not In vitro Aggregation Propensity. Front. Neurosci. 2016, 10, 499. [Google Scholar] [CrossRef]
- Rajpurohit, C.S.; Kumar, V.; Cheffer, A.; Oliveira, D.; Ulrich, H.; Okamoto, O.K.; Zatz, M.; Ansari, U.A.; Khanna, V.K.; Pant, A.B. Mechanistic Insights of Astrocyte-Mediated Hyperactive Autophagy and Loss of Motor Neuron Function in SOD1L39R Linked Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2020, 57, 4117–4133. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Wang, J.; Henderson, M.; Yang, P.; Hagen, B.M.; Siddique, T.; E Vogel, B.; Deng, H.-X.; Fang, S. Nuclear export of misfolded SOD1 mediated by a normally buried NES-like sequence reduces proteotoxicity in the nucleus. eLife 2017, 6, e23759. [Google Scholar] [CrossRef]
- Amendola, L.M.; Jarvik, G.P.; Leo, M.C.; McLaughlin, H.M.; Akkari, Y.; Amaral, M.D.; Berg, J.S.; Biswas, S.; Bowling, K.M.; Conlin, L.K.; et al. Performance of ACMG-AMP Variant-Interpretation Guidelines among Nine Laboratories in the Clinical Sequencing Exploratory Research Consortium. Am. J. Hum. Genet. 2016, 99, 247. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Bozzoni, V.; Pansarasa, O.; Diamanti, L.; Nosari, G.; Cereda, C.; Ceroni, M. Amyotrophic lateral sclerosis and environmental factors. Funct. Neurol. 2016, 31, 7–19. [Google Scholar] [CrossRef]
- Highley, J.; Pons, A.L.; Cooper-Knock, J.; Wharton, S.B.; Ince, P.; Shaw, P.; Wood, J.; Kirby, J. Motor neurone disease/amyotrophic lateral sclerosis associated with intermediate length CAG repeat expansions in Ataxin-2 does not have 1C2-positive polyglutamine inclusions. Neuropathol. Appl. Neurobiol. 2015, 42, 377–389. [Google Scholar] [CrossRef]
- Yousefian-Jazi, A.; Sung, M.K.; Lee, T.; Hong, Y.-H.; Choi, J.K.; Choi, J. Functional fine-mapping of noncoding risk variants in amyotrophic lateral sclerosis utilizing convolutional neural network. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Scarlino, S.; Domi, T.; Pozzi, L.; Romano, A.; Pipitone, G.B.; Falzone, Y.M.; Mosca, L.; Penco, S.; Lunetta, C.; Sansone, V.; et al. Burden of Rare Variants in ALS and Axonal Hereditary Neuropathy Genes Influence Survival in ALS: Insights from a Next Generation Sequencing Study of an Italian ALS Cohort. Int. J. Mol. Sci. 2020, 21, 3346. [Google Scholar] [CrossRef]
- Shibata, N.; Asayama, K.; Hirano, A.; Kobayashi, M. Immunohistochemical Study on Superoxide Dismutases in Spinal Cords from Autopsied Patients with Amyotrophic Lateral Sclerosis. Dev. Neurosci. 1996, 18, 492–498. [Google Scholar] [CrossRef]
- Gruzman, A.; Wood, W.L.; Alpert, E.; Prasad, M.D.; Miller, R.G.; Rothstein, J.D.; Bowser, R.; Hamilton, R.; Wood, T.D.; Cleveland, D.W.; et al. Common molecular signature in SOD1 for both sporadic and familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2007, 104, 12524–12529. [Google Scholar] [CrossRef] [PubMed]
- Denham, N.C.; Pearman, C.M.; Ding, W.Y.; Waktare, J.; Gupta, D.; Snowdon, R.; Hall, M.; Cooper, R.; Modi, S.; Todd, D.; et al. Systematic re-evaluation of SCN5A variants associated with Brugada syndrome. J. Cardiovasc. Electrophysiol. 2018, 30, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Sau, D.; De Biasi, S.; Vitellaro-Zuccarello, L.; Riso, P.; Guarnieri, S.; Porrini, M.; Simeoni, S.; Crippa, V.; Onesto, E.; Palazzolo, I.; et al. Mutation of SOD1 in ALS: A gain of a loss of function. Hum. Mol. Genet. 2007, 16, 1604–1618. [Google Scholar] [CrossRef] [PubMed]
- Saccon, R.A.; Bunton-Stasyshyn, R.K.A.; Fisher, E.M.; Fratta, P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 2013, 136, 2342–2358. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).