
An Expanding Toolkit for Heterochromatin Repair Studies




Abstract
:1. Introduction
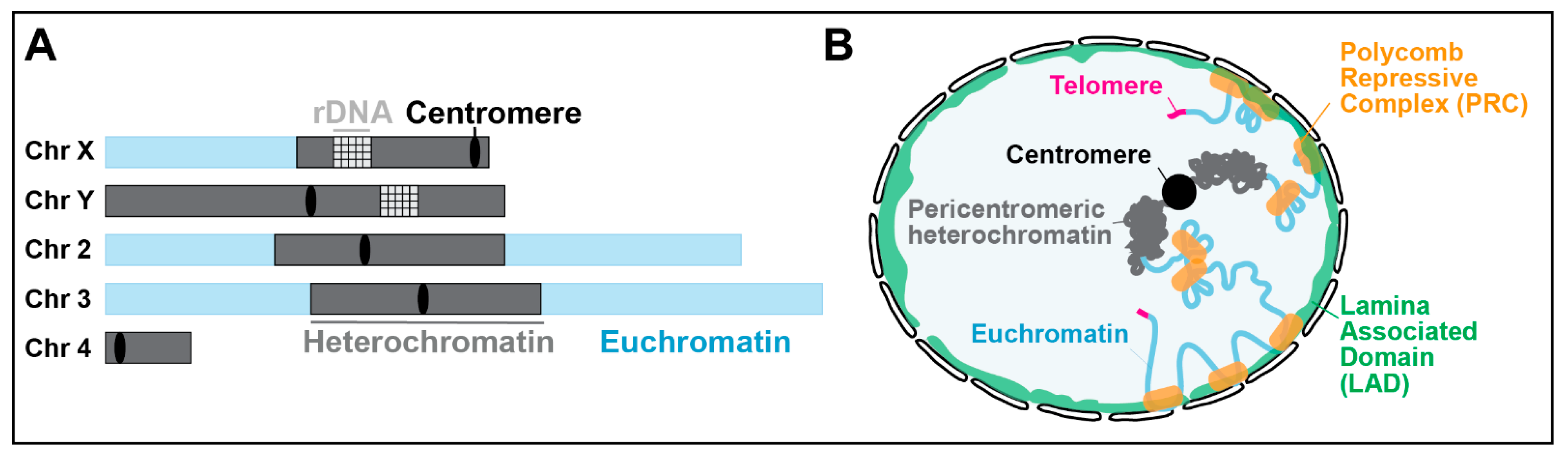
2. Approaches to Study Heterochromatic DSB Responses
2.1. Imaging of Ionizing Radiation (IR)-Induced Repair Foci
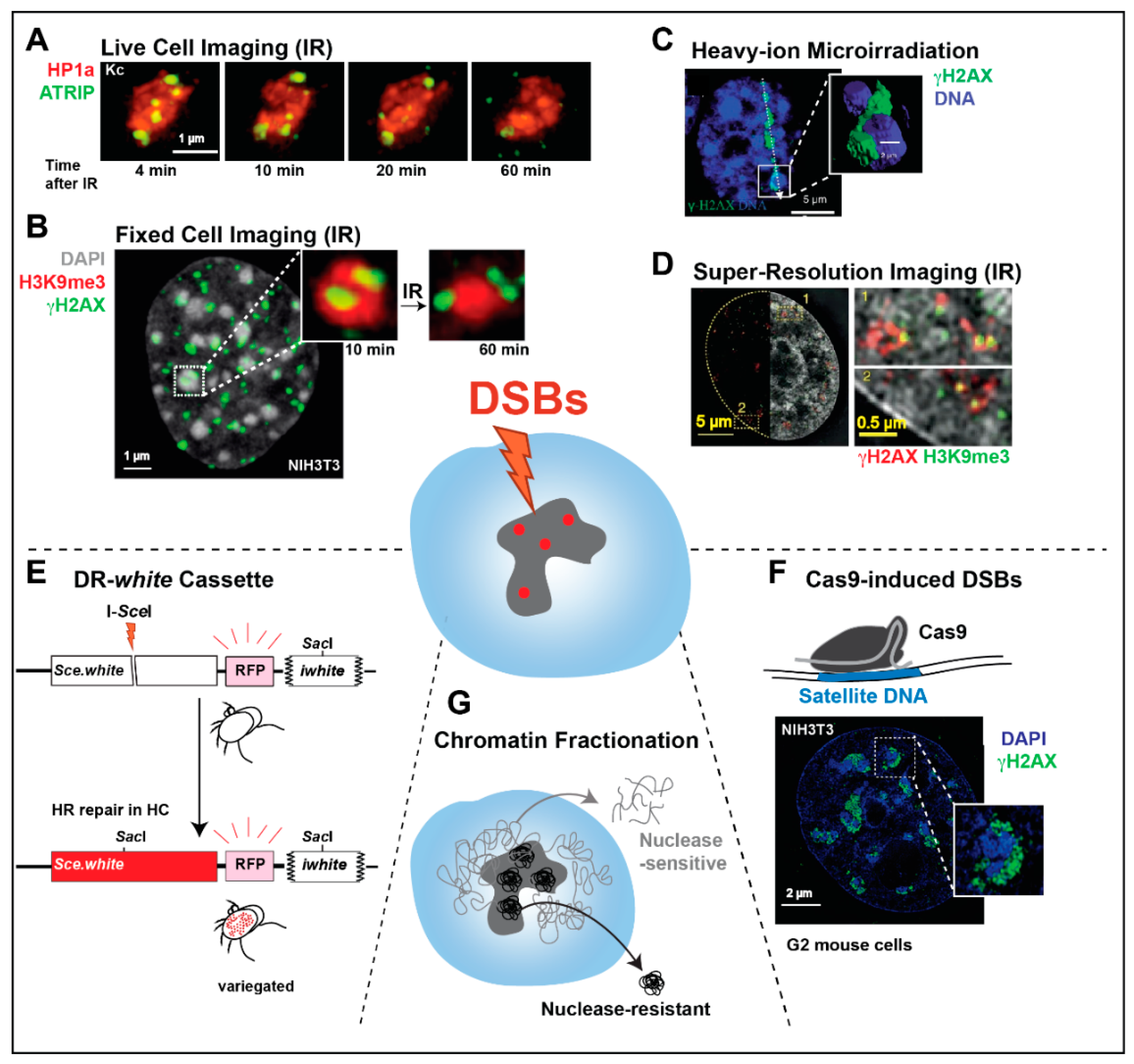
2.2. Imaging of Spatially Defined DSBs Induced by Laser or Heavy-Ion Irradiation
2.3. Super-Resolution Imaging of Repair Responses in Human Heterochromatin
2.4. Chemically Induced DSBs
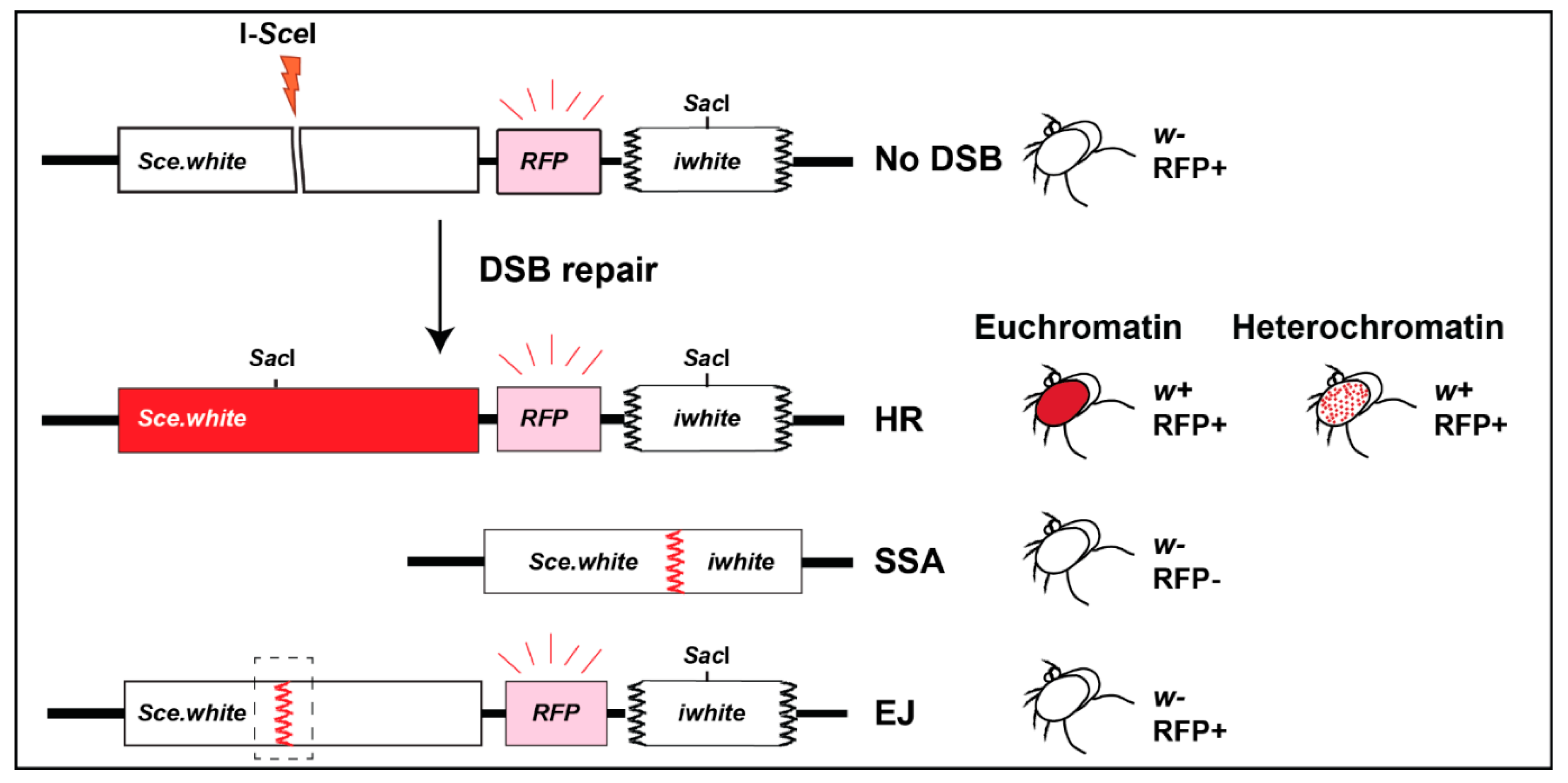
2.5. DR-White Repair Cassette Inserted in Heterochromatin
2.6. Cas9-Induced DSBs in Heterochromatic Satellites
2.7. Biochemical Fractionation of Heterochromatin
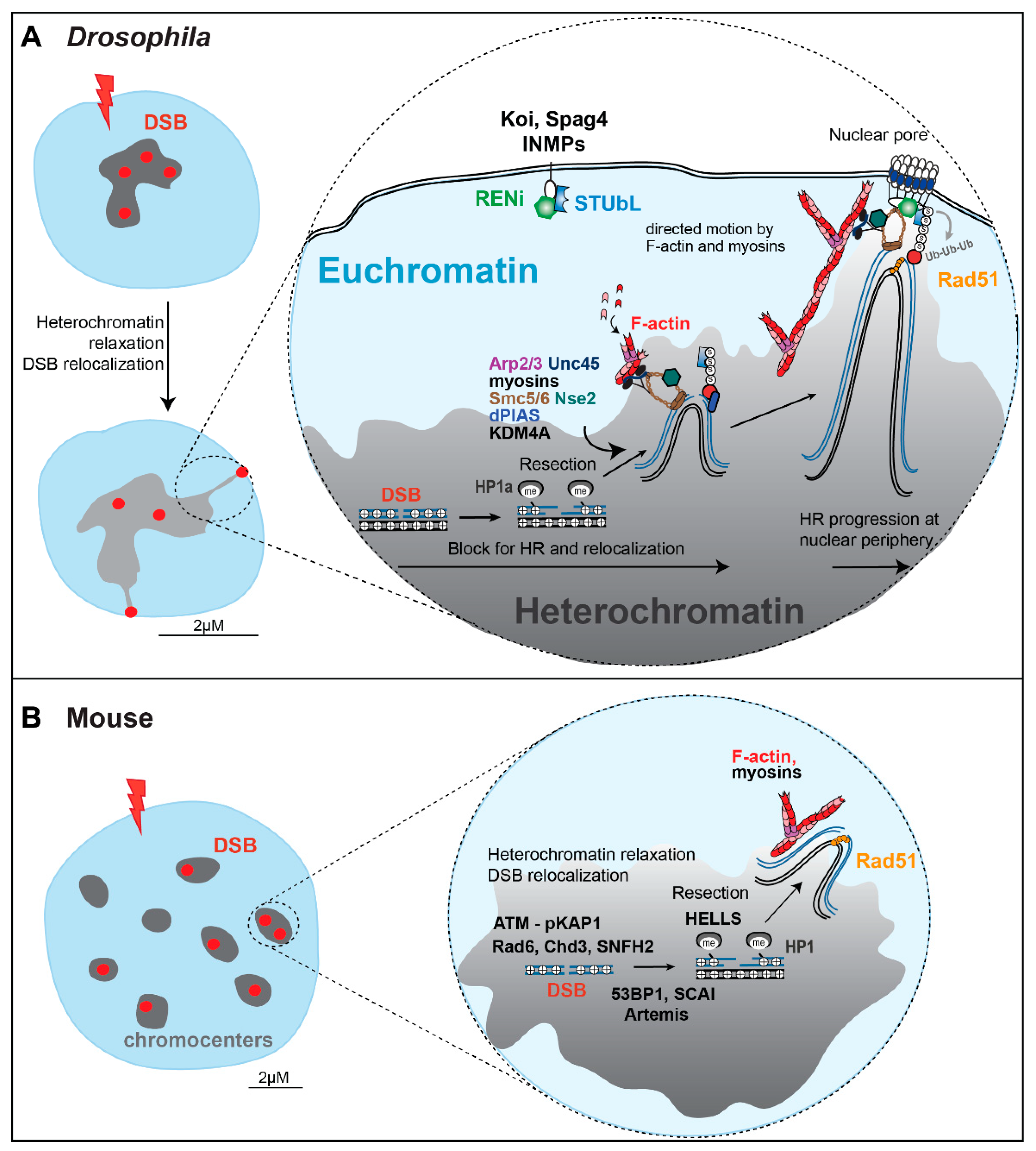
3. Perspectives
3.1. Live and Fixed Cell Imaging
3.2. Site-Specific DSB Induction Systems
3.3. Repair Cassettes
3.4. Exploring the Heterochromatin Damage Proteome with Biochemical Approaches
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Glossary
| Proteins | |
| 53BP1 (P53-binding protein 1) | Promotes DSB repair and checkpoint signaling. |
| Acf1-Snf2h (ATP-dependent chromatin assembly factor complex containing the SWI/SNF related 2h subunit) | Chromatin remodeler |
| Artemis | Structure-specific endonuclease that promotes DSB repair |
| ATM (Ataxia telangiectasia mutated) | DNA damage checkpoint kinase. |
| ATR (Ataxia telangiectasia and Rad3 related) | DNA damage checkpoint kinase. |
| ATRIP (ATR interacting protein) | Protein that mediates ATR recruitment in response to DSBs |
| Brca2 (Breast Cancer 2) | Protein that facilitates Rad51 recruitment onto resected DNA |
| Chd3 (Chromodomain Helicase DNA Binding Protein 3) | Chromatin remodeler. |
| CtIP (C-terminal binding protein (CtBP)-interacting protein | Protein that promotes resection. |
| HELLS (Helicase, Lymphoid Specific) | Chromatin remodeler. |
| HP1 (Heterochromatin protein 1) | Heterochromatin-specific protein associated with H3K9me2/3. |
| KAP1 (Kruppel-associated box (KRAB)-associated protein 1) | Protein that promotes silencing. |
| LINC (Linker of nucleoskeleton and cytoskeleton complex) | Complex spanning the nuclear envelope, composed of a SUN protein across the inner membrane and a KASH protein across the outer membrane. |
| Mdc1 (Mediator of DNA damage checkpoint 1) | Protein that interacts with γH2AX to mediate DSB repair and checkpoint activation. |
| Mu2 (Mutator 2) | Drosophila homologue of Mdc1. |
| Rad54 (Radiation-sensitive mutant protein 54) | Protein that promotes Rad51-mediated strand invasion. |
| Rad6 (Radiation-sensitive mutant protein 6) | E2 ubiquitin-conjugating enzyme that promotes DNA repair progression. |
| RENi (Rad60-Esc2-NIP45 protein family) | Protein family with SUMO-like domains that work in concert with STUbL for DSB repair. |
| RPA (Replication Protein A) | Trimeric complex that binds to ssDNA after resection. |
| SCAI (Suppressor of cancer cell invasion) | Protein that promotes DSB repair. |
| Smc5/6 (Structural maintenance of chromosomes complex 5/6) | Complex that promotes HR. |
| SUMO (Small Ubiquitin-like Modifier) | Peptide covalently attached to other proteins to modify their function. |
| TopBP1 (DNA Topoisomerase II Binding Protein 1) | Protein that promotes ATR activation. |
| Tosca | Drosophila homologue of the exonuclease Exo1 that contributes to resection. |
| Unc45 (Uncoordinated mutant protein 45) | Myosin chaperone and activator. |
| γH2AX/γH2Av | Phosphorylated form of the histone H2A variants H2AX or H2Av. Involved in DSB signaling in mammalian or fly cells, respectively. |
| Methods | |
| 3D-SIM microscopy (3D structured illumination microscopy) | Super resolution microscopy technique. |
| BrdU (5–bromo–2′–deoxyuridine) | Nucleotide analog. |
| CLIP tag | Extrinsically fluorescent protein tag that can be labeled with synthetic probes. |
| CRISPR/Cas9 system (Clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 system) | Technology that can be used to induce site-specific DSBs by the Cas9 endonuclease directed to a sequence of interest. |
| Halo tag | Extrinsically fluorescent protein tag that can be labeled with synthetic probes. |
| Hi-C | Chromatin conformation capture technique used to identify the proximity of genomic regions. |
| I-SceI (Intron-encoded Saccaromyces cerevisiae endonuclease 1) | Endonuclease used to induce site-specific DSBs. |
| MiMIC (Minos-mediated integration cassette) | Collection of insertions of transposon for Drosophila genetic manipulation |
| SNAP tag | Extrinsically fluorescent protein tag that can be labeled with synthetic probes. |
| STED microscopy (Stimulated emission depletion microscopy) | Super resolution microscopy technique. |
| TUNEL (Terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling) | Assay to detect DNA breaks. |
References
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409, 860–921. [Google Scholar] [PubMed] [Green Version]
- Hoskins, R.A.; Carlson, J.W.; Kennedy, C.; Acevedo, D.; Evans-Holm, M.; Frise, E.; Wan, K.H.; Park, S.; Mendez-Lago, M.; Rossi, F.; et al. Sequence Finishing and Mapping of Drosophila Melanogaster Heterochromatin. Science 2007, 316, 1625–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, J.W.K.; Jung, Y.L.; Liu, T.; Alver, B.H.; Lee, S.; Ikegami, K.; Sohn, K.-A.; Minoda, A.; Tolstorukov, M.Y.; Appert, A.; et al. Comparative Analysis of Metazoan Chromatin Organization. Nature 2014, 512, 449–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoskins, R.A.; Carlson, J.W.; Wan, K.H.; Park, S.; Mendez, I.; Galle, S.E.; Booth, B.W.; Pfeiffer, B.D.; George, R.A.; Svirskas, R.; et al. The Release 6 Reference Sequence of the Drosophila Melanogaster Genome. Genome Res. 2015, 25, 445–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riddle, N.C.; Minoda, A.; Kharchenko, P.V.; Alekseyenko, A.A.; Schwartz, Y.B.; Tolstorukov, M.Y.; Gorchakov, A.A.; Jaffe, J.D.; Kennedy, C.; Linder-Basso, D.; et al. Plasticity in Patterns of Histone Modifications and Chromosomal Proteins in Drosophila Heterochromatin. Genome Res. 2011, 21, 147–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawal, C.C.; Caridi, C.P.; Chiolo, I. Actin’ between Phase Separated Domains for Heterochromatin Repair. DNA Repair 2019, 81, 102646. [Google Scholar] [CrossRef] [PubMed]
- Caridi, P.C.; Delabaere, L.; Zapotoczny, G.; Chiolo, I. And Yet, It Moves: Nuclear and Chromatin Dynamics of a Heterochromatic Double-Strand Break. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160291. [Google Scholar] [CrossRef] [Green Version]
- Pickersgill, H.; Kalverda, B.; de Wit, E.; Talhout, W.; Fornerod, M.; van Steensel, B. Characterization of the Drosophila Melanogaster Genome at the Nuclear Lamina. Nat. Genet. 2006, 38, 1005–1014. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Noon, A.T.; Deckbar, D.; Ziv, Y.; Shiloh, Y.; Löbrich, M.; Jeggo, P.A. ATM Signaling Facilitates Repair of DNA Double-Strand Breaks Associated with Heterochromatin. Mol. Cell 2008, 31, 167–177. [Google Scholar] [CrossRef]
- Chiolo, I.; Minoda, A.; Colmenares, S.U.; Polyzos, A.; Costes, S.V.; Karpen, G.H. Double-Strand Breaks in Heterochromatin Move outside of a Dynamic HP1a Domain to Complete Recombinational Repair. Cell 2011, 144, 732–744. [Google Scholar] [CrossRef] [Green Version]
- Chiolo, I.; Tang, J.; Georgescu, W.; Costes, S.V. Nuclear Dynamics of Radiation-Induced Foci in Euchromatin and Heterochromatin. Mutat. Res. 2013, 750, 56–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsouroula, K.; Furst, A.; Rogier, M.; Heyer, V.; Maglott-Roth, A.; Ferrand, A.; Reina-San-Martin, B.; Soutoglou, E. Temporal and Spatial Uncoupling of DNA Double Strand Break Repair Pathways within Mammalian Heterochromatin. Mol. Cell 2016, 63, 293–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjong, H.; Li, W.; Kalhor, R.; Dai, C.; Hao, S.; Gong, K.; Zhou, Y.; Li, H.; Zhou, X.J.; Le Gros, M.A.; et al. Population-Based 3D Genome Structure Analysis Reveals Driving Forces in Spatial Genome Organization. Proc. Natl. Acad. Sci. USA 2016, 113, E1663–E1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Tjong, H.; Li, X.; Gong, K.; Zhou, X.J.; Chiolo, I.; Alber, F. The Three-Dimensional Genome Organization of Drosophila Melanogaster through Data Integration. Genome Biol. 2017, 18, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, T.; Spatola, B.; Delabaere, L.; Bowlin, K.; Hopp, H.; Kunitake, R.; Karpen, G.H.; Chiolo, I. Heterochromatic Breaks Move to the Nuclear Periphery to Continue Recombinational Repair. Nat. Cell Biol. 2015, 17, 1401–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harr, J.C.; Luperchio, T.R.; Wong, X.; Cohen, E.; Wheelan, S.J.; Reddy, K.L. Directed Targeting of Chromatin to the Nuclear Lamina Is Mediated by Chromatin State and A-Type Lamins. J. Cell Biol. 2015, 208, 33–52. [Google Scholar] [CrossRef] [PubMed]
- Briand, N.; Collas, P. Lamina-Associated Domains: Peripheral Matters and Internal Affairs. Genome Biol. 2020, 21, 85. [Google Scholar] [CrossRef] [Green Version]
- Kendek, A.; Wensveen, M.R.; Janssen, A. The Sound of Silence: How Silenced Chromatin Orchestrates the Repair of Double-Strand Breaks. Genes 2021, 12, 1415. [Google Scholar] [CrossRef]
- Peng, J.C.; Karpen, G.H. Epigenetic Regulation of Heterochromatic DNA Stability. Curr. Opin. Genet. Dev. 2008, 18, 204–211. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-Homologous DNA End Joining and Alternative Pathways to Double-Strand Break Repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Cejka, P.; Symington, L.S. DNA End Resection: Mechanism and Control. Annu. Rev. Genet. 2021, 55, 285–307. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.C.; Karpen, G.H. H3K9 Methylation and RNA Interference Regulate Nucleolar Organization and Repeated DNA Stability. Nat. Cell Biol. 2007, 9, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.C.; Karpen, G.H. Heterochromatic Genome Stability Requires Regulators of Histone H3 K9 Methylation. PLoS Genet. 2009, 5, e1000435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, T.; Bonner, M.R.; Chiolo, I. Cervantes and Quijote Protect Heterochromatin from Aberrant Recombination and Lead the Way to the Nuclear Periphery. Nucleus 2016, 7, 485–497. [Google Scholar] [CrossRef] [Green Version]
- Caridi, C.P.; D’Agostino, C.; Ryu, T.; Zapotoczny, G.; Delabaere, L.; Li, X.; Khodaverdian, V.Y.; Amaral, N.; Lin, E.; Rau, A.R.; et al. Nuclear F-Actin and Myosins Drive Relocalization of Heterochromatic Breaks. Nature 2018, 559, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Dialynas, G.; Delabaere, L.; Chiolo, I. Arp2/3 and Unc45 Maintain Heterochromatin Stability in Polytene Chromosomes. Exp. Biol. Med. 2019, 244, 1362–1371. [Google Scholar] [CrossRef]
- Amaral, N.; Ryu, T.; Li, X.; Chiolo, I. Nuclear Dynamics of Heterochromatin Repair. Trends Genet. 2017, 33, 86–100. [Google Scholar] [CrossRef] [Green Version]
- Beucher, A.; Birraux, J.; Tchouandong, L.; Barton, O.; Shibata, A.; Conrad, S.; Goodarzi, A.A.; Krempler, A.; Jeggo, P.A.; Löbrich, M. ATM and Artemis Promote Homologous Recombination of Radiation-Induced DNA Double-Strand Breaks in G2. EMBO J. 2009, 28, 3413–3427. [Google Scholar] [CrossRef] [Green Version]
- Caridi, C.P.; Plessner, M.; Grosse, R.; Chiolo, I. Nuclear Actin Filaments in DNA Repair Dynamics. Nat. Cell Biol. 2019, 21, 1068–1077. [Google Scholar] [CrossRef]
- Miné-Hattab, J.; Chiolo, I. Complex Chromatin Motions for DNA Repair. Front. Genet. 2020, 11, 800. [Google Scholar] [CrossRef]
- Merigliano, C.; Chiolo, I. Multi-Scale Dynamics of Heterochromatin Repair. Curr. Opin. Genet. Dev. 2021, 71, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Jakob, B.; Splinter, J.; Conrad, S.; Voss, K.-O.; Zink, D.; Durante, M.; Löbrich, M.; Taucher-Scholz, G. DNA Double-Strand Breaks in Heterochromatin Elicit Fast Repair Protein Recruitment, Histone H2AX Phosphorylation and Relocation to Euchromatin. Nucleic Acids Res. 2011, 39, 6489–6499. [Google Scholar] [CrossRef]
- Janssen, A.; Breuer, G.A.; Brinkman, E.K.; van der Meulen, A.I.; Borden, S.V.; van Steensel, B.; Bindra, R.S.; LaRocque, J.R.; Karpen, G.H. A Single Double-Strand Break System Reveals Repair Dynamics and Mechanisms in Heterochromatin and Euchromatin. Genes Dev. 2016, 30, 1645–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vítor, A.C.; Huertas, P.; Legube, G.; de Almeida, S.F. Studying DNA Double-Strand Break Repair: An Ever-Growing Toolbox. Front. Mol. Biosci. 2020, 7, 24. [Google Scholar] [CrossRef] [Green Version]
- Mao, P.; Liu, J.; Zhang, Z.; Zhang, H.; Liu, H.; Gao, S.; Rong, Y.S.; Zhao, Y. Homologous Recombination-Dependent Repair of Telomeric DSBs in Proliferating Human Cells. Nat. Commun. 2016, 7, 12154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemaître, C.; Grabarz, A.; Tsouroula, K.; Andronov, L.; Furst, A.; Pankotai, T.; Heyer, V.; Rogier, M.; Attwood, K.M.; Kessler, P.; et al. Nuclear Position Dictates DNA Repair Pathway Choice. Genes Dev. 2014, 28, 2450–2463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schep, R.; Brinkman, E.K.; Leemans, C.; Vergara, X.; van der Weide, R.H.; Morris, B.; van Schaik, T.; Manzo, S.G.; Peric-Hupkes, D.; van den Berg, J.; et al. Impact of Chromatin Context on Cas9-Induced DNA Double-Strand Break Repair Pathway Balance. Mol. Cell 2021, 81, 2216–2230.e10. [Google Scholar] [CrossRef] [PubMed]
- Clouaire, T.; Rocher, V.; Lashgari, A.; Arnould, C.; Aguirrebengoa, M.; Biernacka, A.; Skrzypczak, M.; Aymard, F.; Fongang, B.; Dojer, N.; et al. Comprehensive Mapping of Histone Modifications at DNA Double-Strand Breaks Deciphers Repair Pathway Chromatin Signatures. Mol. Cell 2018, 72, 250–262.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, R.C.; Burman, B.; Kruhlak, M.J.; Misteli, T. Activation of DNA Damage Response Signaling by Condensed Chromatin. Cell Rep. 2014, 9, 1703–1717. [Google Scholar] [CrossRef] [Green Version]
- Aymard, F.; Bugler, B.; Schmidt, C.K.; Guillou, E.; Caron, P.; Briois, S.; Iacovoni, J.S.; Daburon, V.; Miller, K.M.; Jackson, S.P.; et al. Transcriptionally Active Chromatin Recruits Homologous Recombination at DNA Double-Strand Breaks. Nat. Struct. Mol. Biol. 2014, 21, 366–374. [Google Scholar] [CrossRef] [Green Version]
- Fortuny, A.; Chansard, A.; Caron, P.; Chevallier, O.; Leroy, O.; Renaud, O.; Polo, S.E. Imaging the Response to DNA Damage in Heterochromatin Domains Reveals Core Principles of Heterochromatin Maintenance. Nat. Commun. 2021, 12, 2428. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Hale, C.J.; Over, R.S.; Cokus, S.J.; Jacobsen, S.E.; Michaels, S.D. Large-Scale Heterochromatin Remodeling Linked to Overreplication-Associated DNA Damage. Proc. Natl. Acad. Sci. USA 2017, 114, 406–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Yamin, B.; Ahmed-Seghir, S.; Tomida, J.; Despras, E.; Pouvelle, C.; Yurchenko, A.; Goulas, J.; Corre, R.; Delacour, Q.; Droin, N.; et al. DNA Polymerase Zeta Contributes to Heterochromatin Replication to Prevent Genome Instability. EMBO J. 2021, 40, e104543. [Google Scholar] [CrossRef] [PubMed]
- Lisby, M.; Rothstein, R. Choreography of Recombination Proteins during the DNA Damage Response. DNA Repair 2009, 8, 1068–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costes, S.V.; Chiolo, I.; Pluth, J.M.; Barcellos-Hoff, M.H.; Jakob, B. Spatiotemporal Characterization of Ionizing Radiation Induced DNA Damage Foci and Their Relation to Chromatin Organization. Mutat. Res. 2010, 704, 78–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayoub, N.; Jeyasekharan, A.D.; Bernal, J.A.; Venkitaraman, A.R. HP1-β Mobilization Promotes Chromatin Changes That Initiate the DNA Damage Response. Nature 2008, 453, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Jeggo, P.A. The Heterochromatic Barrier to DNA Double Strand Break Repair: How to Get the Entry Visa. Int. J. Mol. Sci. 2012, 13, 11844–11860. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-A.; Kruhlak, M.; Dotiwala, F.; Nussenzweig, A.; Haber, J.E. Heterochromatin Is Refractory to γ-H2AX Modification in Yeast and Mammals. J. Cell Biol. 2007, 178, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Natale, F.; Rapp, A.; Yu, W.; Maiser, A.; Harz, H.; Scholl, A.; Grulich, S.; Anton, T.; Hörl, D.; Chen, W.; et al. Identification of the Elementary Structural Units of the DNA Damage Response. Nat. Commun. 2017, 8, 15760. [Google Scholar] [CrossRef]
- Colmenares, S.U.; Swenson, J.M.; Langley, S.A.; Kennedy, C.; Costes, S.V.; Karpen, G.H. Drosophila Histone Demethylase KDM4A Has Enzymatic and Non-Enzymatic Roles in Controlling Heterochromatin Integrity. Dev. Cell 2017, 42, 156–169.e5. [Google Scholar] [CrossRef] [Green Version]
- See, C.; Arya, D.; Lin, E.; Chiolo, I. Live Cell Imaging of Nuclear Actin Filaments and Heterochromatic Repair Foci in Drosophila and Mouse Cells. Methods Mol. Biol. 2021, 2153, 459–482. [Google Scholar] [PubMed]
- Caridi, C.P.; Delabaere, L.; Tjong, H.; Hopp, H.; Das, D.; Alber, F.; Chiolo, I. Quantitative Methods to Investigate the 4D Dynamics of Heterochromatic Repair Sites in Drosophila Cells. Methods Enzymol. 2018, 601, 359–389. [Google Scholar] [PubMed]
- Klement, K.; Luijsterburg, M.S.; Pinder, J.B.; Cena, C.S.; Del Nero, V.; Wintersinger, C.M.; Dellaire, G.; van Attikum, H.; Goodarzi, A.A. Opposing ISWI- and CHD-Class Chromatin Remodeling Activities Orchestrate Heterochromatic DNA Repair. J. Cell Biol. 2014, 207, 717–733. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Kurka, T.; Jeggo, P.A. KAP-1 Phosphorylation Regulates CHD3 Nucleosome Remodeling during the DNA Double-Strand Break Response. Nat. Struct. Mol. Biol. 2011, 18, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.K.; Mund, A.; Poulsen, S.L.; Sandoval, M.; Klement, K.; Tsouroula, K.; Tollenaere, M.A.X.; Räschle, M.; Soria, R.; Offermanns, S.; et al. SCAI Promotes DNA Double-Strand Break Repair in Distinct Chromosomal Contexts. Nat. Cell Biol. 2016, 18, 1357–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakarougkas, A.; Ismail, A.; Klement, K.; Goodarzi, A.A.; Conrad, S.; Freire, R.; Shibata, A.; Lobrich, M.; Jeggo, P.A. Opposing Roles for 53BP1 during Homologous Recombination. Nucleic Acids Res. 2013, 41, 9719–9731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollárovič, G.; Topping, C.E.; Shaw, E.P.; Chambers, A.L. The Human HELLS Chromatin Remodelling Protein Promotes End Resection to Facilitate Homologous Recombination and Contributes to DSB Repair within Heterochromatin. Nucleic Acids Res. 2020, 48, 1872–1885. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M. Measurement of Radiation-Induced Damage to DNA at the Molecular Level. Int. J. Radiat. Biol. 1992, 61, 175–183. [Google Scholar] [CrossRef]
- Kong, X.; Mohanty, S.K.; Stephens, J.; Heale, J.T.; Gomez-Godinez, V.; Shi, L.Z.; Kim, J.-S.; Yokomori, K.; Berns, M.W. Comparative Analysis of Different Laser Systems to Study Cellular Responses to DNA Damage in Mammalian Cells. Nucleic Acids Res. 2009, 37, e68. [Google Scholar] [CrossRef]
- Mari, P.-O.; Florea, B.I.; Persengiev, S.P.; Verkaik, N.S.; Brüggenwirth, H.T.; Modesti, M.; Giglia-Mari, G.; Bezstarosti, K.; Demmers, J.A.A.; Luider, T.M.; et al. Dynamic Assembly of End-Joining Complexes Requires Interaction between Ku70/80 and XRCC4. Proc. Natl. Acad. Sci. USA 2006, 103, 18597–18602. [Google Scholar] [CrossRef] [Green Version]
- Kruhlak, M.J.; Celeste, A.; Dellaire, G.; Fernandez-Capetillo, O.; Müller, W.G.; McNally, J.G.; Bazett-Jones, D.P.; Nussenzweig, A. Changes in Chromatin Structure and Mobility in Living Cells at Sites of DNA Double-Strand Breaks. J. Cell Biol. 2006, 172, 823–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Yu, Z.; Su, W. Ion irradiation induced direct damage to proteins and their components. arXiv 2008, arXiv:0807.0084v1. [Google Scholar]
- Hada, M.; Georgakilas, A.G. Formation of Clustered DNA Damage after High-LET Irradiation: A Review. J. Radiat. Res. 2008, 49, 203–210. [Google Scholar] [CrossRef]
- Muster, B.; Rapp, A.; Cristina Cardoso, M. Systematic Analysis of DNA Damage Induction and DNA Repair Pathway Activation by Continuous Wave Visible Light Laser Micro-Irradiation. AIMS Genet. 2017, 04, 047–068. [Google Scholar] [CrossRef]
- Timm, S.; Lorat, Y.; Jakob, B.; Taucher-Scholz, G.; Rübe, C.E. Clustered DNA Damage Concentrated in Particle Trajectories Causes Persistent Large-Scale Rearrangements in Chromatin Architecture. Radiother. Oncol. 2018, 129, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.; Stanley, F.K.T.; Goodarzi, A.A. The Repair of Environmentally Relevant DNA Double Strand Breaks Caused by High Linear Energy Transfer Irradiation--No Simple Task. DNA Repair 2014, 17, 64–73. [Google Scholar] [CrossRef]
- Solarczyk, K.J.; Zarębski, M.; Dobrucki, J.W. Inducing Local DNA Damage by Visible Light to Study Chromatin Repair. DNA Repair 2012, 11, 996–1002. [Google Scholar] [CrossRef]
- Dinant, C.; de Jager, M.; Essers, J.; van Cappellen, W.A.; Kanaar, R.; Houtsmuller, A.B.; Vermeulen, W. Activation of Multiple DNA Repair Pathways by Sub-Nuclear Damage Induction Methods. J. Cell Sci. 2007, 120, 2731–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausmann, M.; Wagner, E.; Lee, J.-H.; Schrock, G.; Schaufler, W.; Krufczik, M.; Papenfuß, F.; Port, M.; Bestvater, F.; Scherthan, H. Super-Resolution Localization Microscopy of Radiation-Induced Histone H2AX-Phosphorylation in Relation to H3K9-Trimethylation in HeLa Cells. Nanoscale 2018, 10, 4320–4331. [Google Scholar] [CrossRef]
- Lorković, Z.J.; Park, C.; Goiser, M.; Jiang, D.; Kurzbauer, M.-T.; Schlögelhofer, P.; Berger, F. Compartmentalization of DNA Damage Response between Heterochromatin and Euchromatin Is Mediated by Distinct H2A Histone Variants. Curr. Biol. 2017, 27, 1192–1199. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Ghorai, M.K.; Kenney, G.; Stubbe, J. Mechanistic Studies on Bleomycin-Mediated DNA Damage: Multiple Binding Modes Can Result in Double-Stranded DNA Cleavage. Nucleic Acids Res. 2008, 36, 3781–3790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, A.T.; LaRocque, J.R. The Role of Drosophila Mismatch Repair in Suppressing Recombination between Diverged Sequences. Sci. Rep. 2015, 5, 17601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, K.; McVey, M. Error-Prone Repair of DNA Double-Strand Breaks. J. Cell. Physiol. 2016, 231, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanscom, T.; McVey, M. Regulation of Error-Prone DNA Double-Strand Break Repair and Its Impact on Genome Evolution. Cells 2020, 9, 1657. [Google Scholar] [CrossRef] [PubMed]
- Do, A.T.; Brooks, J.T.; Le Neveu, M.K.; LaRocque, J.R. Double-Strand Break Repair Assays Determine Pathway Choice and Structure of Gene Conversion Events in Drosophila Melanogaster. G3 Genes Genomes Genet. 2014, 4, 425–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, A.; Colmenares, S.U.; Lee, T.; Karpen, G.H. Timely Double-Strand Break Repair and Pathway Choice in Pericentromeric Heterochromatin Depend on the Histone Demethylase dKDM4A. Genes Dev. 2019, 33, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, J.; Bloomer, H.; Kellam, N.; LaRocque, J.R. Chromosome Preference During Homologous Recombination Repair of DNA Double-Strand Breaks in. G3 Genes Genomes Genet. 2019, 9, 3773–3780. [Google Scholar] [CrossRef] [Green Version]
- McKee, B.D. Homologous Pairing and Chromosome Dynamics in Meiosis and Mitosis. Biochim. Biophys. Acta 2004, 1677, 165–180. [Google Scholar] [CrossRef]
- Delabaere, L.; Ertl, H.A.; Massey, D.J.; Hofley, C.M.; Sohail, F.; Bienenstock, E.J.; Sebastian, H.; Chiolo, I.; LaRocque, J.R. Aging Impairs Double-Strand Break Repair by Homologous Recombination in Drosophila Germ Cells. Aging Cell 2017, 16, 320–328. [Google Scholar] [CrossRef]
- Chen, C.-C.; Avdievich, E.; Zhang, Y.; Zhang, Y.; Wei, K.; Lee, K.; Edelmann, W.; Jasin, M.; LaRocque, J.R. EXO1 Suppresses Double-Strand Break Induced Homologous Recombination between Diverged Sequences in Mammalian Cells. DNA Repair 2017, 57, 98–106. [Google Scholar] [CrossRef]
- Ertl, H.A.; Russo, D.P.; Srivastava, N.; Brooks, J.T.; Dao, T.N.; LaRocque, J.R. The Role of Blm Helicase in Homologous Recombination, Gene Conversion Tract Length, and Recombination Between Diverged Sequences in. Genetics 2017, 207, 923–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstock, D.M.; Nakanishi, K.; Helgadottir, H.R.; Jasin, M. Assaying Double-Strand Break Repair Pathway Choice in Mammalian Cells Using a Targeted Endonuclease or the RAG Recombinase. Methods Enzymol. 2006, 409, 524–540. [Google Scholar] [PubMed] [Green Version]
- Gunn, A.; Stark, J.M. I-SceI-Based Assays to Examine Distinct Repair Outcomes of Mammalian Chromosomal Double Strand Breaks. Methods Mol. Biol. 2012, 920, 379–391. [Google Scholar] [PubMed]
- van de Kooij, B.; van Attikum, H. Genomic Reporter Constructs to Monitor Pathway-Specific Repair of DNA Double-Strand Breaks. Front. Genet. 2022, 12, 809832. [Google Scholar] [CrossRef] [PubMed]
- Venken, K.J.T.; Schulze, K.L.; Haelterman, N.A.; Pan, H.; He, Y.; Evans-Holm, M.; Carlson, J.W.; Levis, R.W.; Spradling, A.C.; Hoskins, R.A.; et al. MiMIC: A Highly Versatile Transposon Insertion Resource for Engineering Drosophila Melanogaster Genes. Nat. Methods 2011, 8, 737–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-Guided Genetic Silencing Systems in Bacteria and Archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Mitrentsi, I.; Soutoglou, E. CRISPR/Cas9-Induced Breaks in Heterochromatin, Visualized by Immunofluorescence. Methods Mol. Biol. 2021, 2153, 439–445. [Google Scholar] [PubMed]
- Yilmaz, D.; Furst, A.; Meaburn, K.; Lezaja, A.; Wen, Y.; Altmeyer, M.; Reina-San-Martin, B.; Soutoglou, E. Activation of Homologous Recombination in G1 Preserves Centromeric Integrity. Nature 2021, 600, 748–753. [Google Scholar] [CrossRef]
- van Sluis, M.; McStay, B. A Localized Nucleolar DNA Damage Response Facilitates Recruitment of the Homology-Directed Repair Machinery Independent of Cell Cycle Stage. Genes Dev. 2015, 29, 1151–1163. [Google Scholar] [CrossRef] [Green Version]
- Korsholm, L.M.; Gál, Z.; Lin, L.; Quevedo, O.; Ahmad, D.A.; Dulina, E.; Luo, Y.; Bartek, J.; Larsen, D.H. Double-Strand Breaks in Ribosomal RNA Genes Activate a Distinct Signaling and Chromatin Response to Facilitate Nucleolar Restructuring and Repair. Nucleic Acids Res. 2019, 47, 8019–8035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marnef, A.; Finoux, A.-L.; Arnould, C.; Guillou, E.; Daburon, V.; Rocher, V.; Mangeat, T.; Mangeot, P.E.; Ricci, E.P.; Legube, G. A cohesin/HUSH- and LINC-Dependent Pathway Controls Ribosomal DNA Double-Strand Break Repair. Genes Dev. 2019, 33, 1175–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkman, E.K.; Chen, T.; de Haas, M.; Holland, H.A.; Akhtar, W.; van Steensel, B. Kinetics and Fidelity of the Repair of Cas9-Induced Double-Strand DNA Breaks. Mol. Cell 2018, 70, 801–813.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, C.D.; Kazane, K.R.; Feng, S.J.; Zelin, E.; Bray, N.L.; Schäfer, A.J.; Floor, S.N.; Corn, J.E. CRISPR-Cas9 Genome Editing in Human Cells Occurs via the Fanconi Anemia Pathway. Nat. Genet. 2018, 50, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Mosbach, V.; Viterbo, D.; Descorps-Declère, S.; Poggi, L.; Vaysse-Zinkhöfer, W.; Richard, G.-F. Resection and Repair of a Cas9 Double-Strand Break at CTG Trinucleotide Repeats Induces Local and Extensive Chromosomal Deletions. PLoS Genet. 2020, 16, e1008924. [Google Scholar] [CrossRef] [PubMed]
- Noon, A.T.; Shibata, A.; Rief, N.; Löbrich, M.; Stewart, G.S.; Jeggo, P.A.; Goodarzi, A.A. 53BP1-Dependent Robust Localized KAP-1 Phosphorylation Is Essential for Heterochromatic DNA Double-Strand Break Repair. Nat. Cell Biol. 2010, 12, 177–184. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, X.; Xu, Y.; Ayrapetov, M.K.; Moreau, L.A.; Whetstine, J.R.; Price, B.D. Histone H3 Methylation Links DNA Damage Detection to Activation of the Tumour Suppressor Tip60. Nat. Cell Biol. 2009, 11, 1376–1382. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Wang, C.; Sun, L.; Wang, D.-L.; Chen, L.; Huang, Z.; Yang, Q.; Gao, J.; Yang, X.-B.; Chang, J.-F.; et al. RAD6 Promotes Homologous Recombination Repair by Activating the Autophagy-Mediated Degradation of Heterochromatin Protein HP1. Mol. Cell. Biol. 2015, 35, 406–416. [Google Scholar] [CrossRef] [Green Version]
- Keppler, A.; Gendreizig, S.; Gronemeyer, T.; Pick, H.; Vogel, H.; Johnsson, K. A General Method for the Covalent Labeling of Fusion Proteins with Small Molecules in Vivo. Nat. Biotechnol. 2003, 21, 86–89. [Google Scholar] [CrossRef]
- Los, G.V.; Encell, L.P.; McDougall, M.G.; Hartzell, D.D.; Karassina, N.; Zimprich, C.; Wood, M.G.; Learish, R.; Ohana, R.F.; Urh, M.; et al. HaloTag: A Novel Protein Labeling Technology for Cell Imaging and Protein Analysis. ACS Chem. Biol. 2008, 3, 373–382. [Google Scholar] [CrossRef]
- Gautier, A.; Juillerat, A.; Heinis, C.; Corrêa, I.R., Jr.; Kindermann, M.; Beaufils, F.; Johnsson, K. An Engineered Protein Tag for Multiprotein Labeling in Living Cells. Chem. Biol. 2008, 15, 128–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoelzel, C.A.; Zhang, X. Visualizing and Manipulating Biological Processes by Using HaloTag and SNAP-Tag Technologies. Chembiochem 2020, 21, 1935–1946. [Google Scholar] [CrossRef] [PubMed]
- Jakob, B.; Dubiak-Szepietowska, M.; Janiel, E.; Schmidt, A.; Durante, M.; Taucher-Scholz, G. Differential Repair Protein Recruitment at Sites of Clustered and Isolated DNA Double-Strand Breaks Produced by High-Energy Heavy Ions. Sci. Rep. 2020, 10, 1443. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, W.; Osseiran, A.; Rojec, M.; Liu, Y.; Bombrun, M.; Tang, J.; Costes, S.V. Characterizing the DNA Damage Response by Cell Tracking Algorithms and Cell Features Classification Using High-Content Time-Lapse Analysis. PLoS ONE 2015, 10, e0129438. [Google Scholar]
- Gullberg, M.; Fredriksson, S.; Taussig, M.; Jarvius, J.; Gustafsdottir, S.; Landegren, U. A Sense of Closeness: Protein Detection by Proximity Ligation. Curr. Opin. Biotechnol. 2003, 14, 82–86. [Google Scholar] [CrossRef]
- Strom, A.R.; Emelyanov, A.V.; Mir, M.; Fyodorov, D.V.; Darzacq, X.; Karpen, G.H. Phase Separation Drives Heterochromatin Domain Formation. Nature 2017, 547, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Pessina, F.; Giavazzi, F.; Yin, Y.; Gioia, U.; Vitelli, V.; Galbiati, A.; Barozzi, S.; Garre, M.; Oldani, A.; Flaus, A.; et al. Functional Transcription Promoters at DNA Double-Strand Breaks Mediate RNA-Driven Phase Separation of Damage-Response Factors. Nat. Cell Biol. 2019, 21, 1286–1299. [Google Scholar] [CrossRef] [PubMed]
- Gibson, B.A.; Doolittle, L.K.; Schneider, M.W.G.; Jensen, L.E.; Gamarra, N.; Henry, L.; Gerlich, D.W.; Redding, S.; Rosen, M.K. Organization of Chromatin by Intrinsic and Regulated Phase Separation. Cell 2019, 179, 470–484.e21. [Google Scholar] [CrossRef] [PubMed]
- Kilic, S.; Lezaja, A.; Gatti, M.; Bianco, E.; Michelena, J.; Imhof, R.; Altmeyer, M. Phase Separation of 53BP1 Determines Liquid-like Behavior of DNA Repair Compartments. EMBO J. 2019, 38, e101379. [Google Scholar] [CrossRef] [PubMed]
- Oshidari, R.; Huang, R.; Medghalchi, M.; Tse, E.Y.W.; Ashgriz, N.; Lee, H.O.; Wyatt, H.; Mekhail, K. DNA Repair by Rad52 Liquid Droplets. Nat. Commun. 2020, 11, 695. [Google Scholar] [CrossRef] [Green Version]
- Frattini, C.; Promonet, A.; Alghoul, E.; Vidal-Eychenie, S.; Lamarque, M.; Blanchard, M.-P.; Urbach, S.; Basbous, J.; Constantinou, A. TopBP1 Assembles Nuclear Condensates to Switch on ATR Signaling. Mol. Cell 2021, 81, 1231–1245.e8. [Google Scholar] [CrossRef] [PubMed]
- Spegg, V.; Altmeyer, M. Biomolecular Condensates at Sites of DNA Damage: More than Just a Phase. DNA Repair 2021, 106, 103179. [Google Scholar] [CrossRef] [PubMed]
- Ganser, L.R.; Myong, S. Methods to Study Phase-Separated Condensates and the Underlying Molecular Interactions. Trends Biochem. Sci. 2020, 45, 1004–1005. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Eickbush, D.G.; Speece, I.; Larracuente, A.M. Heterochromatin-Dependent Transcription of Satellite DNAs in the Female Germline. Elife 2021, 10, e62375. [Google Scholar] [CrossRef] [PubMed]
- d’Adda di Fagagna, F. A Direct Role for Small Non-Coding RNAs in DNA Damage Response. Trends Cell Biol. 2014, 24, 171–178. [Google Scholar] [CrossRef]
- Michelini, F.; Jalihal, A.P.; Francia, S.; Meers, C.; Neeb, Z.T.; Rossiello, F.; Gioia, U.; Aguado, J.; Jones-Weinert, C.; Luke, B.; et al. From “Cellular” RNA to “Smart” RNA: Multiple Roles of RNA in Genome Stability and Beyond. Chem. Rev. 2018, 118, 4365–4403. [Google Scholar] [CrossRef]
- Pessina, F.; Gioia, U.; Brandi, O.; Farina, S.; Ceccon, M.; Francia, S.; d’Adda di Fagagna, F. DNA Damage Triggers a New Phase in Neurodegeneration. Trends Genet. 2021, 37, 337–354. [Google Scholar] [CrossRef] [PubMed]
- Rossiello, F.; Aguado, J.; Sepe, S.; Iannelli, F.; Nguyen, Q.; Pitchiaya, S.; Carninci, P.; d’Adda di Fagagna, F. DNA Damage Response Inhibition at Dysfunctional Telomeres by Modulation of Telomeric DNA Damage Response RNAs. Nat. Commun. 2017, 8, 13980. [Google Scholar] [CrossRef] [Green Version]
- Michelini, F.; Pitchiaya, S.; Vitelli, V.; Sharma, S.; Gioia, U.; Pessina, F.; Cabrini, M.; Wang, Y.; Capozzo, I.; Iannelli, F.; et al. Damage-Induced lncRNAs Control the DNA Damage Response through Interaction with DDRNAs at Individual Double-Strand Breaks. Nat. Cell Biol. 2017, 19, 1400–1411. [Google Scholar] [CrossRef] [Green Version]
- Aguado, J.; Sola-Carvajal, A.; Cancila, V.; Revêchon, G.; Ong, P.F.; Jones-Weinert, C.W.; Arzt, E.W.; Lattanzi, G.; Dreesen, O.; Tripodo, C.; et al. Inhibition of DNA Damage Response at Telomeres Improves the Detrimental Phenotypes of Hutchinson–Gilford Progeria Syndrome. Nat. Commun. 2019, 10, 4990. [Google Scholar] [CrossRef]
- Iacovoni, J.S.; Caron, P.; Lassadi, I.; Nicolas, E.; Massip, L.; Trouche, D.; Legube, G. High-Resolution Profiling of gammaH2AX around DNA Double Strand Breaks in the Mammalian Genome. EMBO J. 2010, 29, 1446–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Caron, P.; Legube, G.; Paull, T.T. Quantitation of DNA Double-Strand Break Resection Intermediates in Human Cells. Nucleic Acids Res. 2014, 42, e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canela, A.; Sridharan, S.; Sciascia, N.; Tubbs, A.; Meltzer, P.; Sleckman, B.P.; Nussenzweig, A. DNA Breaks and End Resection Measured Genome-Wide by End Sequencing. Mol. Cell 2016, 63, 898–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, N.; John, S.; Nussenzweig, A.; Canela, A. END-Seq: An Unbiased, High-Resolution, and Genome-Wide Approach to Map DNA Double-Strand Breaks and Resection in Human Cells. Methods Mol. Biol. 2021, 2153, 9–31. [Google Scholar] [PubMed]
- Clouaire, T.; Legube, G. DNA Double Strand Break Repair Pathway Choice: A Chromatin Based Decision? Nucleus 2015, 6, 107–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnould, C.; Rocher, V.; Finoux, A.-L.; Clouaire, T.; Li, K.; Zhou, F.; Caron, P.; Mangeot, P.E.; Ricci, E.P.; Mourad, R.; et al. Loop Extrusion as a Mechanism for Formation of DNA Damage Repair Foci. Nature 2021, 590, 660–665. [Google Scholar] [CrossRef]
- Lechner, M.S.; Schultz, D.C.; Negorev, D.; Maul, G.G.; Rauscher, F.J., 3rd. The Mammalian Heterochromatin Protein 1 Binds Diverse Nuclear Proteins through a Common Motif That Targets the Chromoshadow Domain. Biochem. Biophys. Res. Commun. 2005, 331, 929–937. [Google Scholar] [CrossRef]
- Rosnoblet, C.; Vandamme, J.; Völkel, P.; Angrand, P.-O. Analysis of the Human HP1 Interactome Reveals Novel Binding Partners. Biochem. Biophys. Res. Commun. 2011, 413, 206–211. [Google Scholar] [CrossRef]
- Motamedi, M.R.; Hong, E.-J.E.; Li, X.; Gerber, S.; Denison, C.; Gygi, S.; Moazed, D. HP1 Proteins Form Distinct Complexes and Mediate Heterochromatic Gene Silencing by Nonoverlapping Mechanisms. Mol. Cell 2008, 32, 778–790. [Google Scholar] [CrossRef] [Green Version]
- Iglesias, N.; Paulo, J.A.; Tatarakis, A.; Wang, X.; Edwards, A.L.; Bhanu, N.V.; Garcia, B.A.; Haas, W.; Gygi, S.P.; Moazed, D. Native Chromatin Proteomics Reveals a Role for Specific Nucleoporins in Heterochromatin Organization and Maintenance. Mol. Cell 2020, 77, 51–66.e8. [Google Scholar] [CrossRef]
- Alekseyenko, A.A.; Gorchakov, A.A.; Zee, B.M.; Fuchs, S.M.; Kharchenko, P.V.; Kuroda, M.I. Heterochromatin-Associated Interactions of Drosophila HP1a with dADD1, HIPP1, and Repetitive RNAs. Genes Dev. 2014, 28, 1445–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, H.-W.; Lee, D.H.; Florens, L.; Swanson, S.K.; Washburn, M.P.; Kwon, S.H. Analysis of the Heterochromatin Protein 1 (HP1) Interactome in Drosophila. J. Proteom. 2014, 102, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A Promiscuous Biotin Ligase Fusion Protein Identifies Proximal and Interacting Proteins in Mammalian Cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Chojnowski, A.; Ong, P.F.; Wong, E.S.M.; Lim, J.S.Y.; Mutalif, R.A.; Navasankari, R.; Dutta, B.; Yang, H.; Liow, Y.Y.; Sze, S.K.; et al. Progerin Reduces LAP2α-Telomere Association in Hutchinson-Gilford Progeria. Elife 2015, 4, e07759. [Google Scholar] [CrossRef] [PubMed]
- Schmidtmann, E.; Anton, T.; Rombaut, P.; Herzog, F.; Leonhardt, H. Determination of Local Chromatin Composition by CasID. Nucleus 2016, 7, 476–484. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Meng, Y.; Wang, Y.; Li, J.; Lam, M.; Wang, L.; Di, L.-J. Nuclear Localization of Desmoplakin and Its Involvement in Telomere Maintenance. Int. J. Biol. Sci. 2019, 15, 2350–2362. [Google Scholar] [CrossRef] [PubMed]
- Santos-Barriopedro, I.; van Mierlo, G.; Vermeulen, M. Off-the-Shelf Proximity Biotinylation for Interaction Proteomics. Nat. Commun. 2021, 12, 5015. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.S.; McCarthy, R.L.; Sidoli, S.; Donahue, G.; Kaeding, K.E.; He, Z.; Lin, S.; Garcia, B.A.; Zaret, K.S. Genomic and Proteomic Resolution of Heterochromatin and Its Restriction of Alternate Fate Genes. Mol. Cell 2017, 68, 1023–1037.e15. [Google Scholar] [CrossRef] [Green Version]
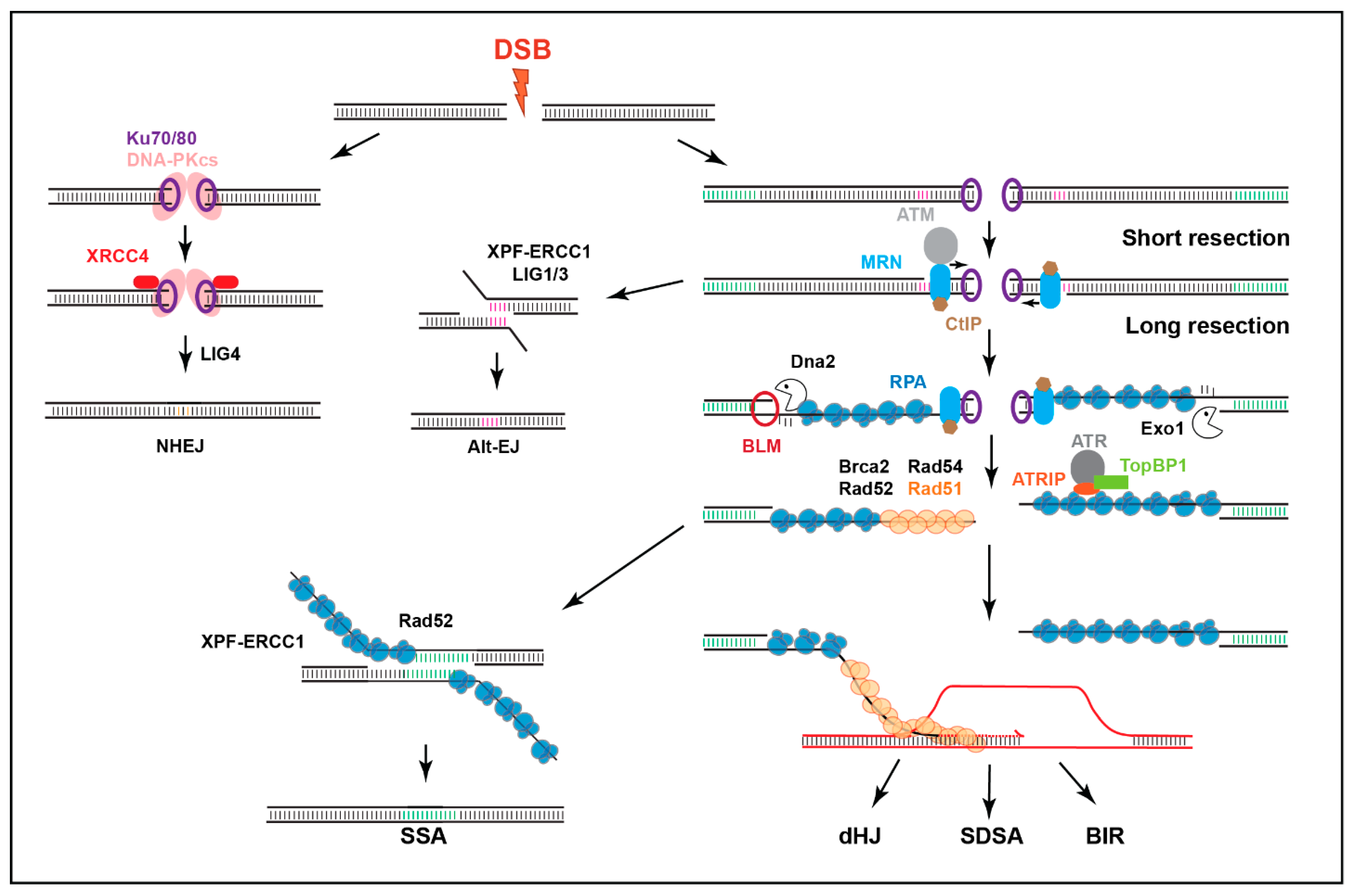

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique Applied to Heterochromatin | Advantages | Disadvantages | Refs. |
|---|---|---|---|
| Imaging of IR-induced foci |
|
| [9,10,15,25,26,28,50,51,52,53,54,55,56,57] |
| Imaging after laser or heavy-ion irradiation |
|
| [10,32,46] |
| Super-resolution imaging |
|
| [49,69] |
| Chemically induced DSBs |
|
| [70] |
| DR-white repair cassette |
|
| [33,76] |
| Cas9-induced DSBs in satellites. |
|
| [12,76,88] |
| Biochemical fractionation |
|
| [9,53,96,98] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rawal, C.C.; Butova, N.L.; Mitra, A.; Chiolo, I. An Expanding Toolkit for Heterochromatin Repair Studies. Genes 2022, 13, 529. https://doi.org/10.3390/genes13030529
Rawal CC, Butova NL, Mitra A, Chiolo I. An Expanding Toolkit for Heterochromatin Repair Studies. Genes. 2022; 13(3):529. https://doi.org/10.3390/genes13030529
Chicago/Turabian StyleRawal, Chetan C., Nadejda L. Butova, Anik Mitra, and Irene Chiolo. 2022. "An Expanding Toolkit for Heterochromatin Repair Studies" Genes 13, no. 3: 529. https://doi.org/10.3390/genes13030529
APA StyleRawal, C. C., Butova, N. L., Mitra, A., & Chiolo, I. (2022). An Expanding Toolkit for Heterochromatin Repair Studies. Genes, 13(3), 529. https://doi.org/10.3390/genes13030529