
First Description of Inheritance of a Postzygotic OPA1 Mosaic Variant

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
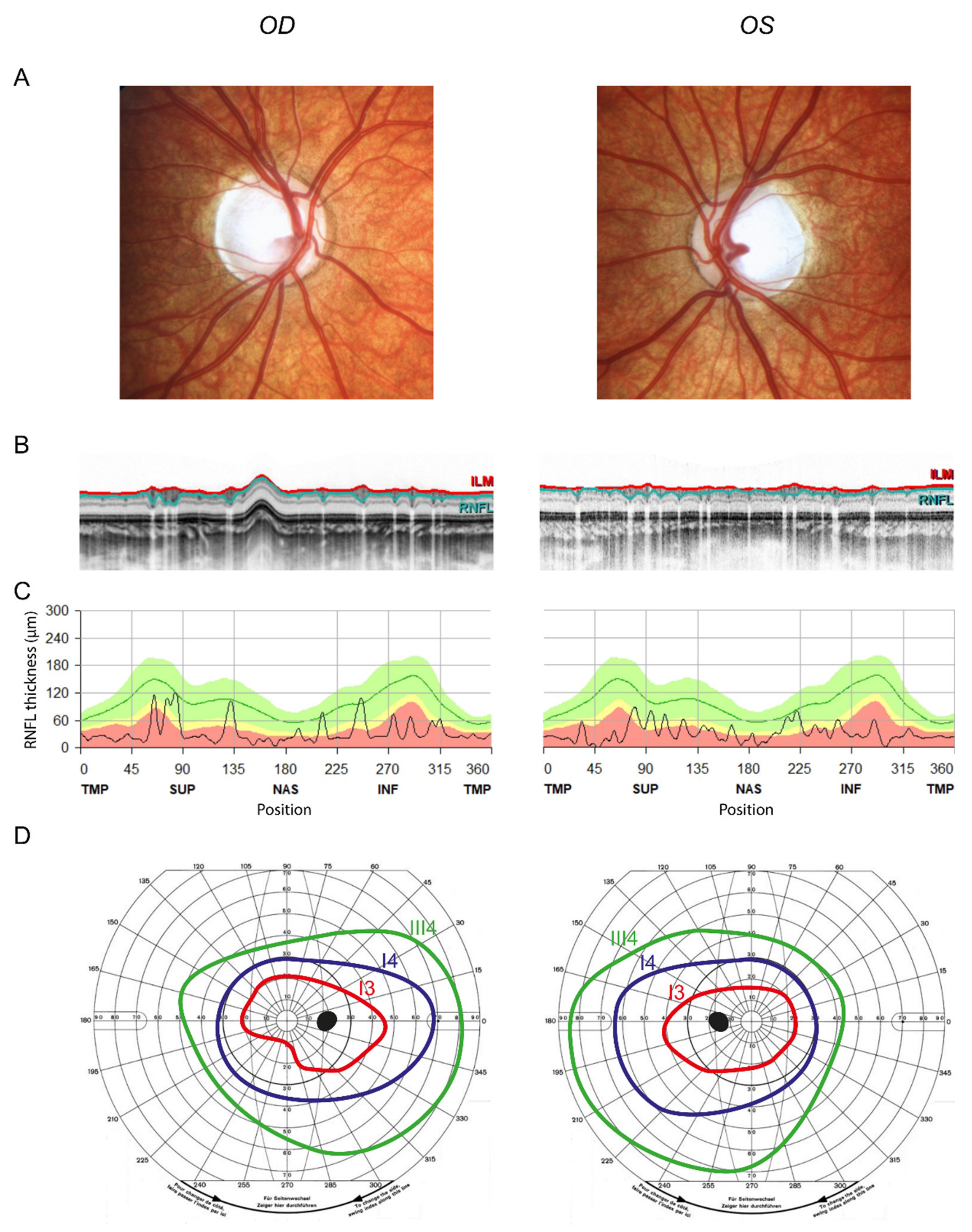
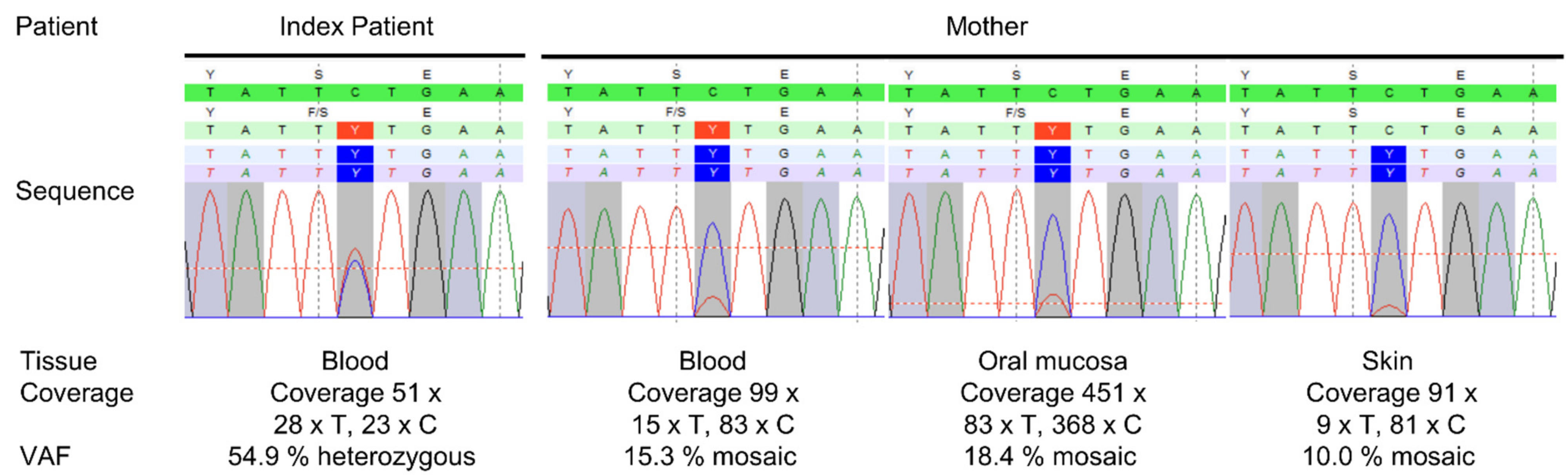
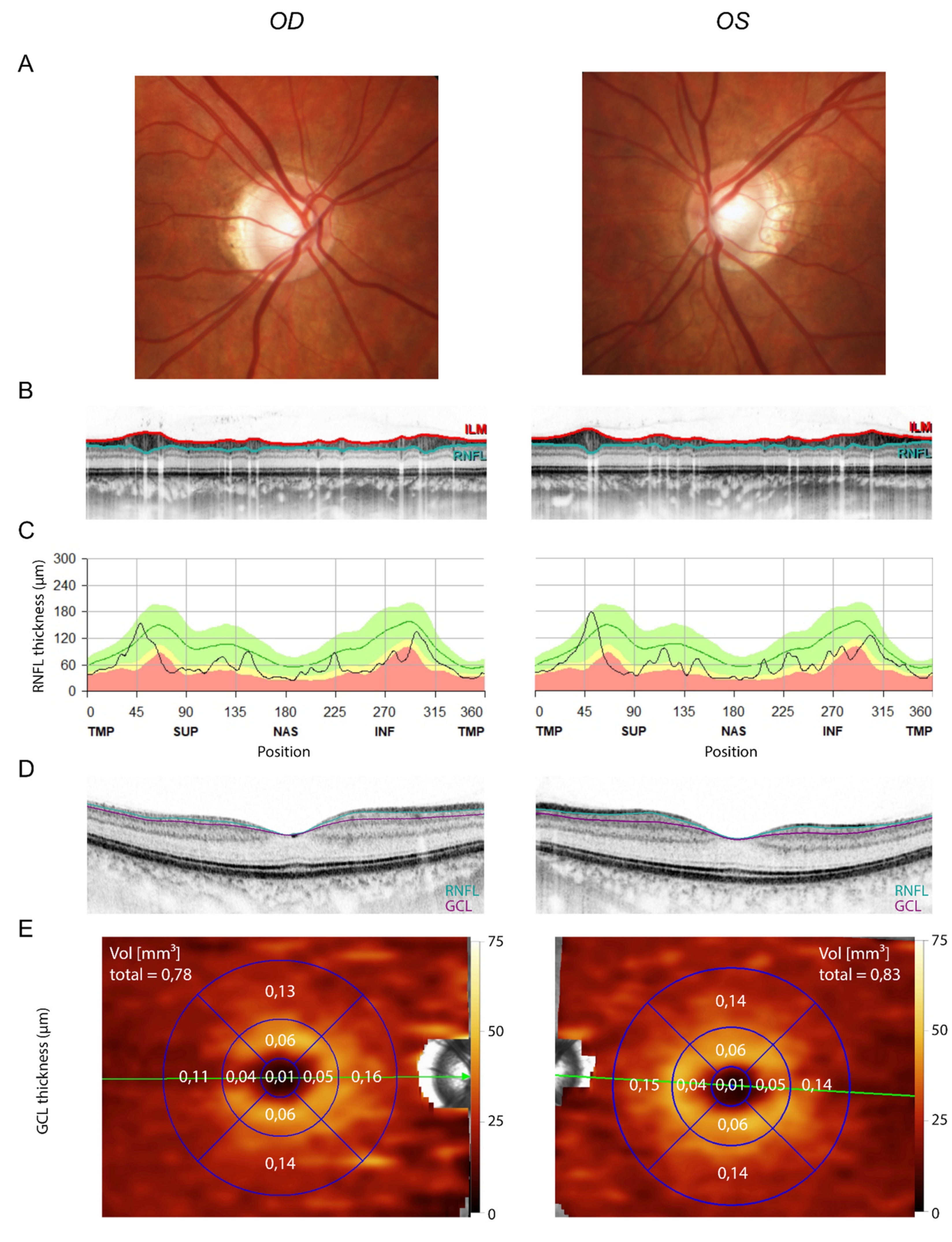
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferré, M.; Bonneau, D.; Milea, D.; Chevrollier, A.; Verny, C.; Dollfus, H.; Ayuso, C.; Defoort, S.; Vignal, C.; Zanlonghi, X.; et al. Molecular Screening of 980 Cases of Suspected Hereditary Optic Neuropathy with a Report on 77 Novel OPA1 Mutations. Hum. Mutat. 2009, 30, E692–E705. [Google Scholar] [CrossRef] [PubMed]
- Newman, N.J. Hereditary Optic Neuropathies: From the Mitochondria to the Optic Nerve. Am. J. Ophthalmol. 2005, 140, 517.e1–517.e9. [Google Scholar] [CrossRef] [PubMed]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 Requires Mitofusin 1 to Promote Mitochondrial Fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 Controls Apoptotic Cristae Remodeling Independently from Mitochondrial Fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Trenell, M.I.; Hollingsworth, K.G.; Griffiths, P.G.; Chinnery, P.F. OPA1 Mutations Impair Mitochondrial Function in Both Pure and Complicated Dominant Optic Atrophy. Brain 2011, 134, e164. [Google Scholar] [CrossRef] [PubMed]
- Lenaers, G.; Reynier, P.; Elachouri, G.; Soukkarieh, C.; Olichon, A.; Belenguer, P.; Baricault, L.; Ducommun, B.; Hamel, C.; Delettre, C. OPA1 Functions in Mitochondria and Dysfunctions in Optic Nerve. Int. J. Biochem. Cell Biol. 2009, 41, 1866–1874. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 Processing Controls Mitochondrial Fusion and Is Regulated by MRNA Splicing, Membrane Potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef]
- Delettre, C.; Lenaers, G.; Pelloquin, L.; Belenguer, P.; Hamel, C.P. OPA1 (Kjer Type) Dominant Optic Atrophy: A Novel Mitochondrial Disease. Mol. Genet. Metab. 2002, 75, 97–107. [Google Scholar] [CrossRef]
- Skidd, P.M.; Lessell, S.; Cestari, D.M. Autosomal Dominant Hereditary Optic Neuropathy (ADOA): A Review of the Genetics and Clinical Manifestations of ADOA and ADOA+. Semin. Ophthalmol. 2013, 28, 422–426. [Google Scholar] [CrossRef]
- Hoyt, C.S. Autosomal Dominant Optic Atrophy. Ophthalmology 1980, 87, 245–251. [Google Scholar] [CrossRef]
- Cohn, A.C.; Toomes, C.; Potter, C.; Towns, K.V.; Hewitt, A.W.; Inglehearn, C.F.; Craig, J.E.; Mackey, D.A. Autosomal Dominant Optic Atrophy: Penetrance and Expressivity in Patients with OPA1 Mutations. Am. J. Ophthalmol. 2007, 143, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Cohn, A.C.; Toomes, C.; Hewitt, A.W.; Kearns, L.S.; Inglehearn, C.F.; Craig, J.E.; Mackey, D.A. The Natural History of OPA1-Related Autosomal Dominant Optic Atrophy. Br. J. Ophthalmol. 2008, 92, 1333–1336. [Google Scholar] [CrossRef] [PubMed]
- Votruba, M.; Moore, A.T.; Bhattacharya, S.S. Clinical Features, Molecular Genetics, and Pathophysiology of Dominant Optic Atrophy. J. Med. Genet. 1998, 35, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Griffiths, P.G.; Gorman, G.S.; Lourenco, C.M.; Wright, A.F.; Auer-Grumbach, M.; Toscano, A.; Musumeci, O.; Valentino, M.L.; Caporali, L.; et al. Multi-System Neurological Disease Is Common in Patients with OPA1 Mutations. Brain 2010, 133, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, D.; Colin, E.; Oca, F.; Ferré, M.; Chevrollier, A.; Guéguen, N.; Desquiret-Dumas, V.; N’Guyen, S.; Barth, M.; Zanlonghi, X.; et al. Early-Onset Behr Syndrome Due to Compound Heterozygous Mutations in OPA1. Brain 2014, 137, e301. [Google Scholar] [CrossRef]
- Barboni, P.; Valentino, M.L.; La Morgia, C.; Carbonelli, M.; Savini, G.; De Negri, A.; Simonelli, F.; Sadun, F.; Caporali, L.; Maresca, A.; et al. Idebenone Treatment in Patients with OPA1-Mutant Dominant Optic Atrophy. Brain 2013, 136, e231. [Google Scholar] [CrossRef]
- Delettre-Cribaillet, C.; Hamel, C.P.; Lenaers, G. Optic Atrophy Type 1. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Acuna-Hidalgo, R.; Veltman, J.A.; Hoischen, A. New Insights into the Generation and Role of de Novo Mutations in Health and Disease. Genome Biol. 2016, 17, 241. [Google Scholar] [CrossRef]
- Baris, O.; Delettre, C.; Amati-Bonneau, P.; Surget, M.-O.; Charlin, J.-F.; Catier, A.; Derieux, L.; Guyomard, J.-L.; Dollfus, H.; Jonveaux, P.; et al. Fourteen Novel OPA1 Mutations in Autosomal Dominant Optic Atrophy Including Two de Novo Mutations in Sporadic Optic Atrophy. Hum. Mutat. 2003, 21, 656. [Google Scholar] [CrossRef]
- Cohen, L.; Tzur, S.; Goldenberg-Cohen, N.; Bormans, C.; Behar, D.M.; Reinstein, E. Exome Sequencing Identified a Novel de Novo OPA1 Mutation in a Consanguineous Family Presenting with Optic Atrophy. Genet. Res. 2016, 98, e10. [Google Scholar] [CrossRef]
- Le Roux, B.; Lenaers, G.; Zanlonghi, X.; Amati-Bonneau, P.; Chabrun, F.; Foulonneau, T.; Caignard, A.; Leruez, S.; Gohier, P.; Procaccio, V.; et al. OPA1: 516 Unique Variants and 831 Patients Registered in an Updated Centralized Variome Database. Orphanet J. Rare Dis. 2019, 14, 214. [Google Scholar] [CrossRef]
- Chen, J.; Xu, K.; Zhang, X.; Jiang, F.; Liu, L.; Dong, B.; Ren, Y.; Li, Y. Mutation Screening of Mitochondrial DNA as Well as OPA1 and OPA3 in a Chinese Cohort with Suspected Hereditary Optic Atrophy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6987–6995. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pesch, U.E.; Leo-Kottler, B.; Mayer, S.; Jurklies, B.; Kellner, U.; Apfelstedt-Sylla, E.; Zrenner, E.; Alexander, C.; Wissinger, B. OPA1 Mutations in Patients with Autosomal Dominant Optic Atrophy and Evidence for Semi-Dominant Inheritance. Hum. Mol. Genet. 2001, 10, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Weisschuh, N.; Schimpf-Linzenbold, S.; Mazzola, P.; Kieninger, S.; Xiao, T.; Kellner, U.; Neuhann, T.; Kelbsch, C.; Tonagel, F.; Wilhelm, H.; et al. Mutation Spectrum of the OPA1 Gene in a Large Cohort of Patients with Suspected Dominant Optic Atrophy: Identification and Classification of 48 Novel Variants. PLoS ONE 2021, 16, e0253987. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.; Karri, R.; Danesh-Meyer, H.; Drummond, K.; Symons, A. A Normative Database of A-Scan Data Using the Heidelberg Spectralis Spectral Domain Optical Coherence Tomography Machine. PLoS ONE 2021, 16, e0253720. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Bae, T.; Tomasini, L.; Mariani, J.; Zhou, B.; Roychowdhury, T.; Franjic, D.; Pletikos, M.; Pattni, R.; Chen, B.-J.; Venturini, E.; et al. Different Mutational Rates and Mechanisms in Human Cells at Pregastrulation and Neurogenesis. Science 2018, 359, 550–555. [Google Scholar] [CrossRef]
- Moog, U.; Felbor, U.; Has, C.; Zirn, B. Disorders Caused by Genetic Mosaicism. Dtsch. Arztebl. Int. 2020, 116, 119–125. [Google Scholar] [CrossRef]
- Tai, E.L.M.; Ling, J.L.; Gan, E.H.; Adil, H.; Wan-Hazabbah, W.-H. Comparison of Peripapillary Retinal Nerve Fiber Layer Thickness between Myopia Severity Groups and Controls. Int. J. Ophthalmol. 2018, 11, 274–278. [Google Scholar] [CrossRef]
- Kang, S.H.; Hong, S.W.; Im, S.K.; Lee, S.H.; Ahn, M.D. Effect of Myopia on the Thickness of the Retinal Nerve Fiber Layer Measured by Cirrus HD Optical Coherence Tomography. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4075–4083. [Google Scholar] [CrossRef]
- Aydogan, T.; Akçay, B.İ.S.; Kardeş, E.; Ergin, A. Evaluation of Spectral Domain Optical Coherence Tomography Parameters in Ocular Hypertension, Preperimetric, and Early Glaucoma. Indian J. Ophthalmol. 2017, 65, 1143–1150. [Google Scholar] [CrossRef]
- Savini, G.; Barboni, P.; Parisi, V.; Carbonelli, M. The Influence of Axial Length on Retinal Nerve Fibre Layer Thickness and Optic-Disc Size Measurements by Spectral-Domain OCT. Br. J. Ophthalmol. 2012, 96, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.M.; Sparkes, R.S. Segmental Neurofibromatosis. Arch. Dermatol. 1977, 113, 837–838. [Google Scholar] [CrossRef] [PubMed]
- Tinschert, S.; Naumann, I.; Stegmann, E.; Buske, A.; Kaufmann, D.; Thiel, G.; Jenne, D.E. Segmental Neurofibromatosis Is Caused by Somatic Mutation of the Neurofibromatosis Type 1 (NF1) Gene. Eur. J. Hum. Genet. 2000, 8, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Chia, S.Y.; Tan, E.-C.; Wei, H.; Zhao, Y.; Koh, M.J.A. Epidermolytic Ichthyosis in a Child and Systematized Epidermolytic Nevi in the Mosaic Parent Associated with a KRT1 Variant. Eur. J. Med. Genet. 2021, 64, 104324. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alter, S.; Farassat, N.; Küchlin, S.; Lagrèze, W.A.; Fischer, J. First Description of Inheritance of a Postzygotic OPA1 Mosaic Variant. Genes 2022, 13, 478. https://doi.org/10.3390/genes13030478
Alter S, Farassat N, Küchlin S, Lagrèze WA, Fischer J. First Description of Inheritance of a Postzygotic OPA1 Mosaic Variant. Genes. 2022; 13(3):478. https://doi.org/10.3390/genes13030478
Chicago/Turabian StyleAlter, Svenja, Navid Farassat, Sebastian Küchlin, Wolf A. Lagrèze, and Judith Fischer. 2022. "First Description of Inheritance of a Postzygotic OPA1 Mosaic Variant" Genes 13, no. 3: 478. https://doi.org/10.3390/genes13030478
APA StyleAlter, S., Farassat, N., Küchlin, S., Lagrèze, W. A., & Fischer, J. (2022). First Description of Inheritance of a Postzygotic OPA1 Mosaic Variant. Genes, 13(3), 478. https://doi.org/10.3390/genes13030478