Abstract
Gemcitabine is a nucleoside analog that has been used widely as an anticancer drug for the treatment of a variety of conditions, including ovarian, bladder, non-small-cell lung, pancreatic, and breast cancer. However, enzymatic deamination, fast systemic clearance, and the emergence of chemoresistance have limited its efficacy. Different prodrug strategies have been explored in recent years, seeking to obtain better pharmacokinetic properties, efficacy, and safety. Different drug delivery strategies have also been employed, seeking to transform gemcitabine into a targeted medicine. This review will provide an overview of the recent developments in gemcitabine prodrugs and their effectiveness in treating cancerous tumors.
1. Introduction
Gemcitabine (GCB) is a nucleoside analog that has been widely used as an antimetabolite antineoplastic agent. GCB has been approved by the US FDA since 1996, and has been sold under the brand name Gemzar. It is administered alone or in combination with other anticancer drugs to treat a variety of conditions, including ovarian, bladder, non-small-cell lung, pancreatic, and breast cancer [1,2]. At the cellular level, GCB is internalized via nucleic acid transporters. It is subsequently phosphorylated by dioxycytidine kinase (DCK). The stepwise phosphorylation leads to the formation of GCB-triphosphate, which is incorporated into cellular DNA, thereby inhibiting nuclear replication.
The clinical efficacy of GCB is affected by a number of well-documented limitations. Enzymatic deamination by cytidine deaminase (CDA) can lead to the formation of inactive 2′-deoxy-2′,2′-difluorouridine (dFdU) [3]. Cancer cells can acquire chemoresistance by the decreased expression of nucleoside transporters, especially the human equilibrative nucleoside transporter (hENT) [4]. Poor tumor targeting can lead to undesirable adverse effects. To address these challenges, multiple attempts have been made to develop prodrugs of GCB that would be stable during enzymatic inactivation, have improved pharmacokinetic properties, and could be employed for targeted cancer therapy. This review will describe the chemical strategies that have been reported in recent years.
2. Prodrugs of Gemcitabine
2.1. Hydrophobic Prodrugs with Different Mechanisms of Cellular Uptake
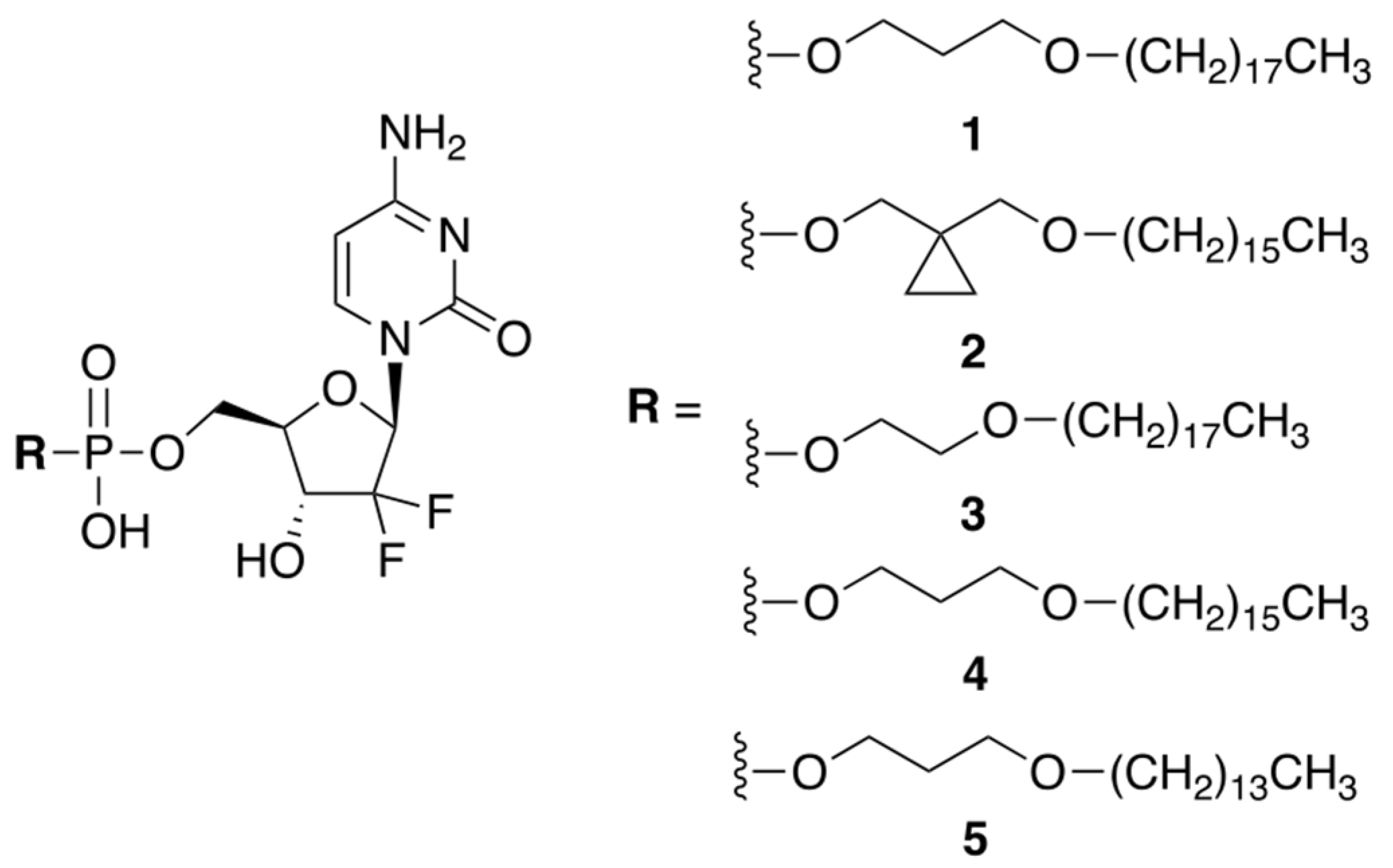
Huixin Qi et al. reported a small library of prodrug compounds, shown in Figure 1, that contain hydrophobic monophosphate ester modifications at the 5′-position of GCB [5]. In the prodrug form, these compounds were expected to be cell permeable, aided by their long hydrophobic tails. Unexpectedly, the reported compounds were found to enter the cells via a different mechanism than GCB. It has previously been determined that GCB utilizes nucleic acid transporters, such as hENT, to achieve cell permeability. The authors showed that inhibiting hENT using dipyridamole did not significantly impact cellular uptake of the prodrugs, suggesting that they likely enter the cell by passive diffusion through the cellular membrane. This is potentially advantageous, as downregulation of hENT can cause cellular chemoresistance to GCB [6]. Inside the cell, the prodrugs were expected to be enzymatically converted to the 5′-monophosphate of GCB and, ultimately, incorporated into the cellular DNA. The reported compounds were evaluated in vitro against a variety of cancer cells using an MTT assay. Prodrug 3 showed the best potency, which was comparable to the cytotoxicity of GCB, especially in A549, DU145, and PC-3 cell lines.
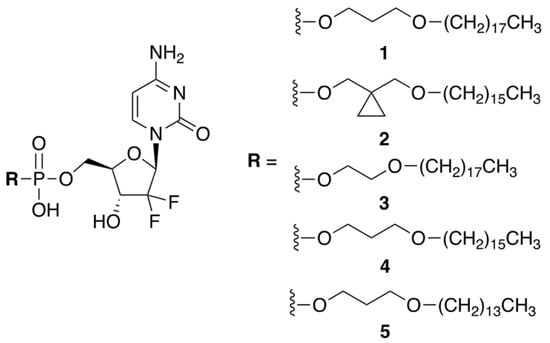
Figure 1.
A library of prodrugs of GCB containing hydrophobic monophosphate ester modifications.
The hydrophobic tail of prodrug 3 reduced its solubility in aqueous media. To achieve therapeutically meaningful doses, the prodrug had to be formulated in a saline solution containing 1% CMC-Na, with 0.5% Tween 80. Pharmacokinetic parameters were evaluated in nude mice after oral administration of prodrug 3 and GCB. The prodrug dose of 20 mg/kg (9.4 mg/kg GCB equiv.) delivered similar systemic exposure as compared to the parent drug. The in vivo efficacy was investigated using a H460 tumor xenograft model (non-small-cell lung cancer). Four oral prodrug doses of 40 mg/kg (18.7 mg/kg GCB equiv.), given once every 3 days, resulted in 65.2% tumor suppression. By comparison, four IP GCB doses of 80 mg/kg, given once every 3 days, resulted in 61.1% tumor suppression. Thus, lower doses of the prodrug were able to achieve comparable efficacy to GCB.
2.2. Prodrugs That Respond to Enzymatic Activation
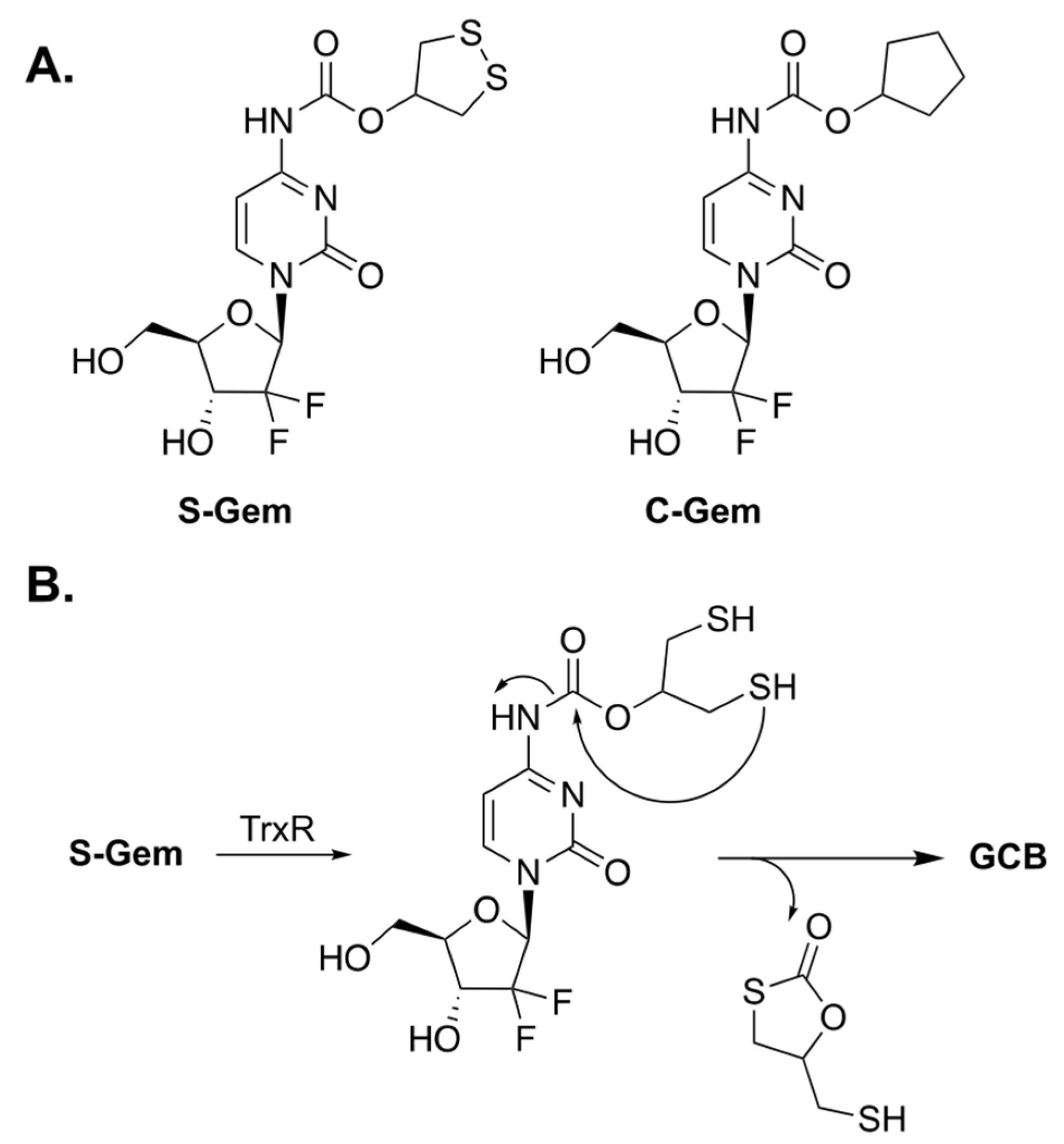
Xinming Li et al. designed a prodrug of GCB, termed S-Gem (Figure 2A), whose enzymatic activation can be triggered by thioredoxin reductase (TrxR) [7]. Malignant cancer cells have abnormally high levels of TrxR for maintaining their tumor phenotypes [8]; thus, it was hypothesized that prodrug activation would be selectively achieved inside the cancer cells. The key residue of TrxR is selenocysteine (Sec). It is an analogue of cysteine in which the sulfur atom is replaced by selenium. A Sec residue has a significantly lower pKa value than that of the thiol group in Cys (5.8 for Sec and 8.3 for Cys), which renders the selenol group in Sec to be predominantly present as selenolate. The authors claim that S-Gem activation is drastically lower if the Sec residue of TrxR is replaced by Cys. The proposed mechanism of activation of S-Gem by TrxR is illustrated in Figure 2B. The enzyme reduces the disulfide bond of 1,2-dithiolane, which leads to intramolecular cyclization and subsequent activation of GCB.
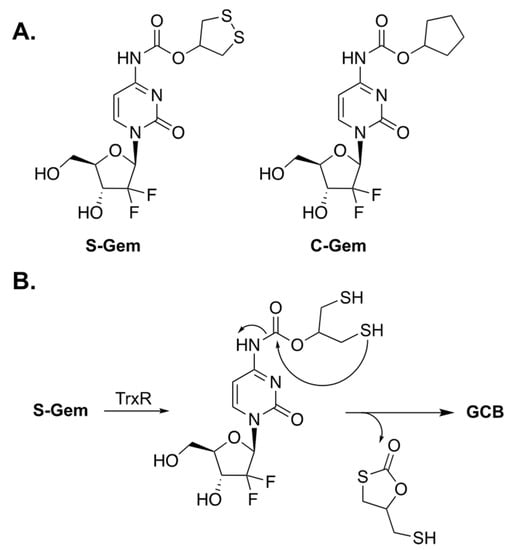
Figure 2.
(A) Prodrugs of GCB, S-Gem and C-Gem. (B) Mechanism of enzymatic activation of S-Gem.
To understand the mechanism of prodrug activation, the authors carried out HPLC studies. Incubation of S-Gem in a buffered media containing TrxR resulted in about 80% release of activated GCB after 4 h. The authors synthesized a negative control analog of the prodrug, in which both the sulfur atoms were replaced by carbons (C-Gem). Incubation of C-Gem with the artificial reducing agent TCEP (tris(2-carboxyethyl)phosphine) produced a small amount of activated GCB. Meanwhile, the incubation of S-Gem with TCEP resulted in a nearly quantitative amount of GCB. Interestingly, the authors also reported that reduced TrxR is capable of activating S-Gem to produce GCB. However, this unexpected finding was not investigated any further. Subsequently, prodrug activation was studied in HeLa cells, which express TrxR. Prodrug activation has been observed in HeLa cell lysates. However, inhibiting the enzyme with auranofin considerably decreased prodrug activation.
The cytotoxicity studies of S-Gem and C-Gem (negative control) were carried out in the following three cell lines: SMMC-7721 cells (human hepatocellular carcinoma), A549 cells (adenocarcinomic human alveolar basal epithelial cells), and HeLa cells (human cervical cancer cells). MTT studies determined that the IC50 values were 1.4 μM for SMMC-7721, 0.6 μM for A549, and 2.2 μM for HeLa cells. Meanwhile, C-Gem was virtually non-toxic. To confirm that the cytotoxicity of S-Gem is dependent on TrxR, the authors performed MTT studies using HeLa cells in which the enzyme was knocked down. Consistent with the proposed hypothesis, the knock down of TrxR resulted in, roughly, a 10-fold increase in the observed IC50 value.
The authors did not evaluate their prodrug in vivo. It would be very interesting to investigate the prodrug’s in vivo behavior. A number of critical elements would be at play. Firstly, the solubility of the prodrug is likely to be lower than that of GCB, which is administered as an HCl salt. Secondly, the prodrug will likely be more lipophilic, which will affect its pharmacokinetic properties. Lastly, the stability of the carbamate group in blood plasma will be highly important.
2.3. Prodrugs That Are Activated by ROS
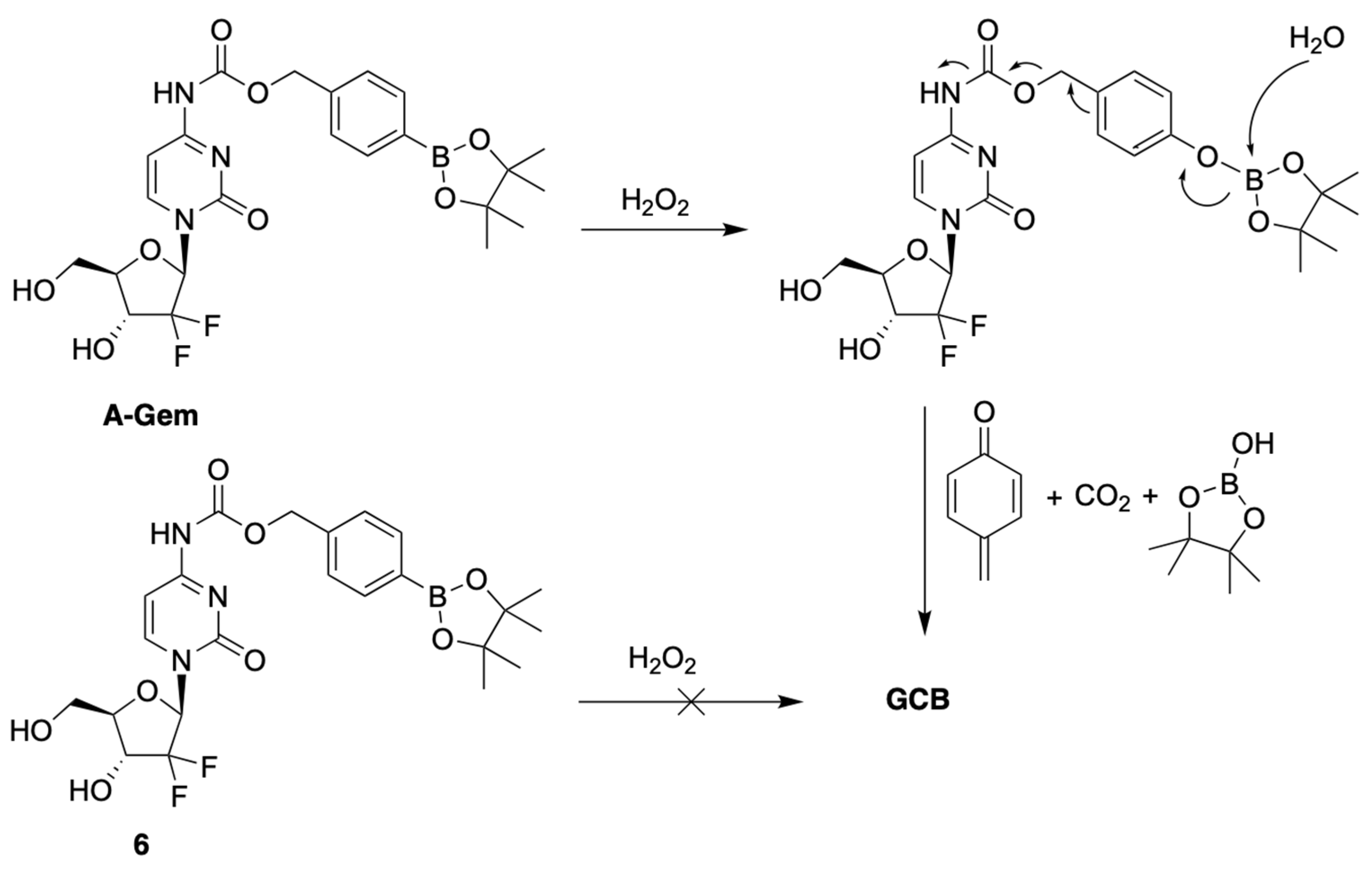
Matsushita et al. developed a prodrug of GCB, termed A-Gem (Figure 3), which releases the active anticancer drug in the presence of H2O2 [9]. Oxidative stress is a common feature of many cancers, resulting in elevated levels of reactive oxygen species (ROS) compared to normal cells [10,11,12,13]. H2O2 is an example of ROS, which is known for its cell permeability and stability [14,15,16,17]. The reported prodrug design is based on the following well-established chemistry: reduction of alkyl and arylboronic acids by H2O2 [18]. This reaction is known to be bio-orthogonal and biocompatible. The prodrug A-Gem follows the same reaction mechanism, as it contains a boronate-ester carbamate group at the N4-position of the nucleobase (Figure 3). The authors also reported a structurally similar negative control compound, 6, which does not have the boronate-ester linkage (Figure 3).
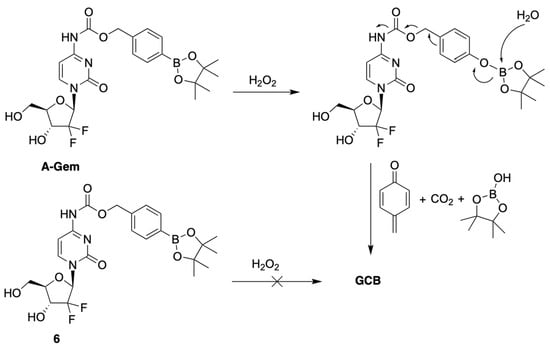
Figure 3.
Mechanism of activation of A-Gem by hydrogen peroxide.
The authors evaluated the mechanism behind the uncaging of A-Gem by H2O2 using HPLC analysis. When A-Gem was treated with an equimolar amount of H2O2, under simulated physiological conditions, GCB was observed within 20 min, with 95% conversion. On the other hand, 6 was not converted into GCB under the same treatment. Interestingly, the activation of A-Gem was shown to be highly selective for H2O2 compared to other ROS present in the human body, such as tert-butylhydroperoxide (TBHP), hypochlorite (ClO−), hydroxyl radical (OH−), tert-butoxy radical (tBuO−), nitric oxide (NO), and superoxide (O2−).
The investigation of the cytotoxicity of GCB, A-Gem, and 6 was carried out in human pancreatic cancer cell lines (PSN1 and BxPC3) and a normal pancreatic epithelial cell line (NPEC), using an MTT assay. As anticipated, the cytotoxicity of 6 was considerably lower that of A-Gem in the cancer cells. On the other hand, their cytotoxicity in NPEC cells, which have lower levels of H2O2, was essentially the same. The addition of exogenous H2O2 resulted in higher cytotoxicity of A-Gem, thus indicating that the tested cancer cells do not have a sufficiently high endogenous concentration of H2O2 to efficiently activate all of the dosed prodrug.
The in vivo evaluation of therapeutic efficacy was carried out in the following three cohorts of mice: GCB, A-Gem, and the vehicle. The A-Gem group received a two-times higher dose (100 mg/kg) than the GCB group (50 mg/kg). The mice were dosed four times with GCB, A-Gem, and the vehicle on days 14, 17, 20, and 23. The prodrug had statistically analogous efficacy to GCB. The average tumor suppression was essentially the same in the A-Gem and GCB cohorts. On the other hand, myelosuppression, the most common side effect of GCB therapy, was lower in the prodrug cohort. LC-MS analysis of the tumor tissue showed similar levels of the activated drug in the A-Gem and GCB cohorts. On the other hand, LC-MS analysis of the bone marrow indicated lower amounts of the activated drug in the prodrug cohort. These findings support the hypothesis that A-Gem behaves as a tumor-selective anticancer agent.
2.4. Prodrugs That Attempt to Counter Multidrug Resistance
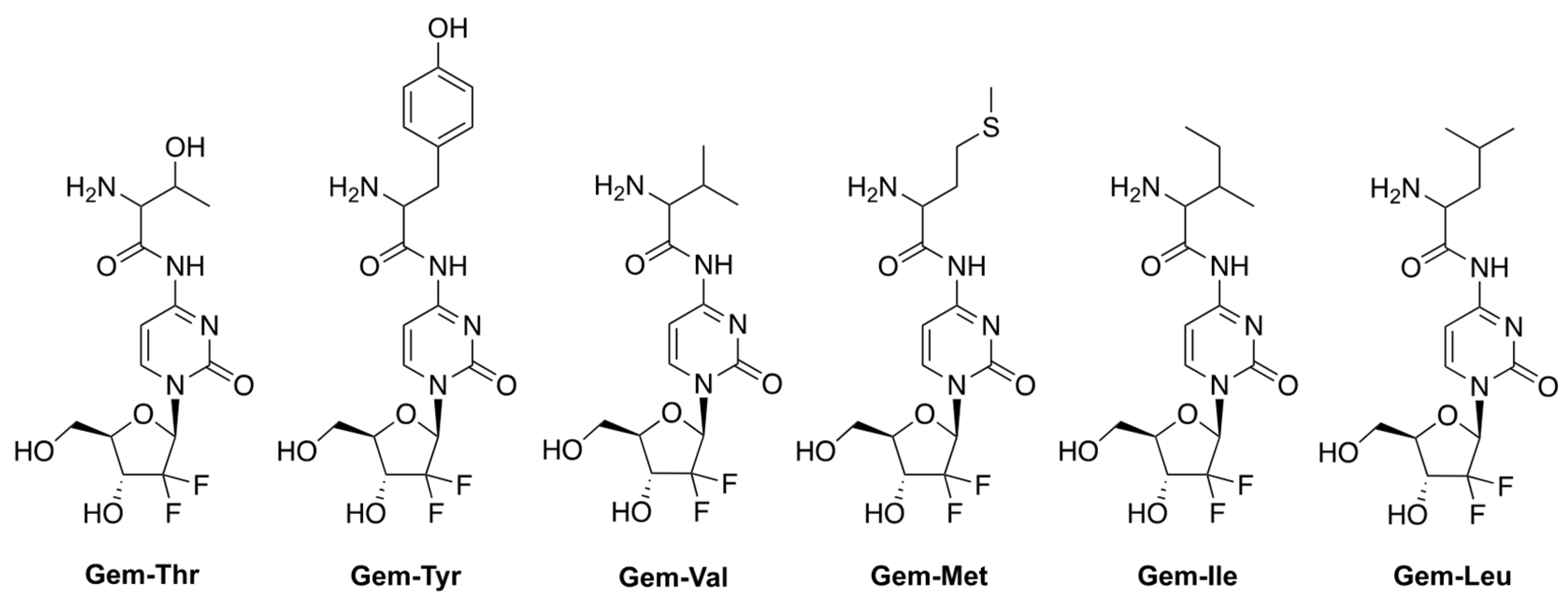
Hong et al. reported prodrugs of GCB that contain various amino acids conjugated to the N4-position of the nucleobase of GCB (Figure 4) [19]. This strategy aims to address one of the mechanisms of multidrug resistance (MDR) triggered by the overexpression of various efflux transporters by cancer cells [20,21,22]. The strategy takes advantage of the concomitant overexpression of amino acid uptake transporters by cancer cells undergoing neoplastic transformation [23]. The authors, in fact, confirmed that the levels of the amino acid transporter LAT-1 were elevated in the pancreatic cancer cell lines BxPC-3 and MIAPaCa-2. The LAT-1 transporter is persistently found in patients with pancreatic cancer, and in transplanted Colo357 cells (pancreatic cancer cell line) [24,25]. The authors hypothesized that using an amino acid-conjugated prodrug of GCB might be an effective way to improve GCB uptake via transporters, such as LAT-1. A threonine derivative of GCB (Gem-Thr) would be transported inside the cancer cells be activated by LAT-1 via amide bond cleavage.
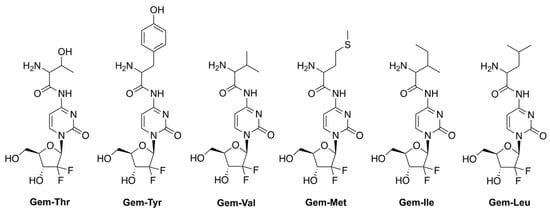
Figure 4.
Prodrugs of GCB, containing various amino acids conjugated to the N4-position of the nucleobase.
The authors investigated the viability of various cancer cells treated with 1 mM prodrugs for 48 h. Gem-Thr showed a similar profile to GCB, being the most cytotoxic against BxPC-3 and B16 cell lines. On the other hand, it was mostly non-toxic against MIAPaCa-2, A549, and MDA-231 cells. The authors performed a TUNEL assay, which suggested that apoptosis was induced by GCB and Gem-Thr treatments.
The stability of Gem-Thr was investigated in PBS, rat blood plasma, and liver microsomes using LC-MS. After incubation for 8 h at 37 °C, the prodrug was found to be remarkably metabolically stable in PBS and rat liver microsomes, even in the presence of elevated cytochrome P450. In rat blood plasma, 20% of Gem-Thr was converted to GCB.
The pharmacokinetics parameters of GCB and Gem-Thr were investigated in rats by IV administration of a 4 mg/kg dose. The AUC and the measure of drug elimination from the body (CL) were found to be 948.38 ± 52.04 μg·min/mL and 4.23 ± 0.23 mL/min/kg, respectively. The volume of distribution at steady state (Vss) and the average time for a drug molecule to reside in the body (MRT), for GCB, were 2483.64 ± 867.19 mL/kg and 582.06 ± 177.90 min, respectively. After administration of Gem-Thr at 4 mg/kg IV, the conversion of Gem-Thr to GCB was found to be likely due to amide bond cleavage. More importantly, Gem-Thr increased systemic exposure (i.e., as defined by the AUC of GCB) by 1.83-fold versus free GCB. This was attributed to the significantly lower total CL value (0.60 vs. 4.23 mL/min/kg). These findings suggest that the amide prodrug approach improves metabolic stability in vivo.
2.5. Macromolecular Prodrugs of Gemcitabine
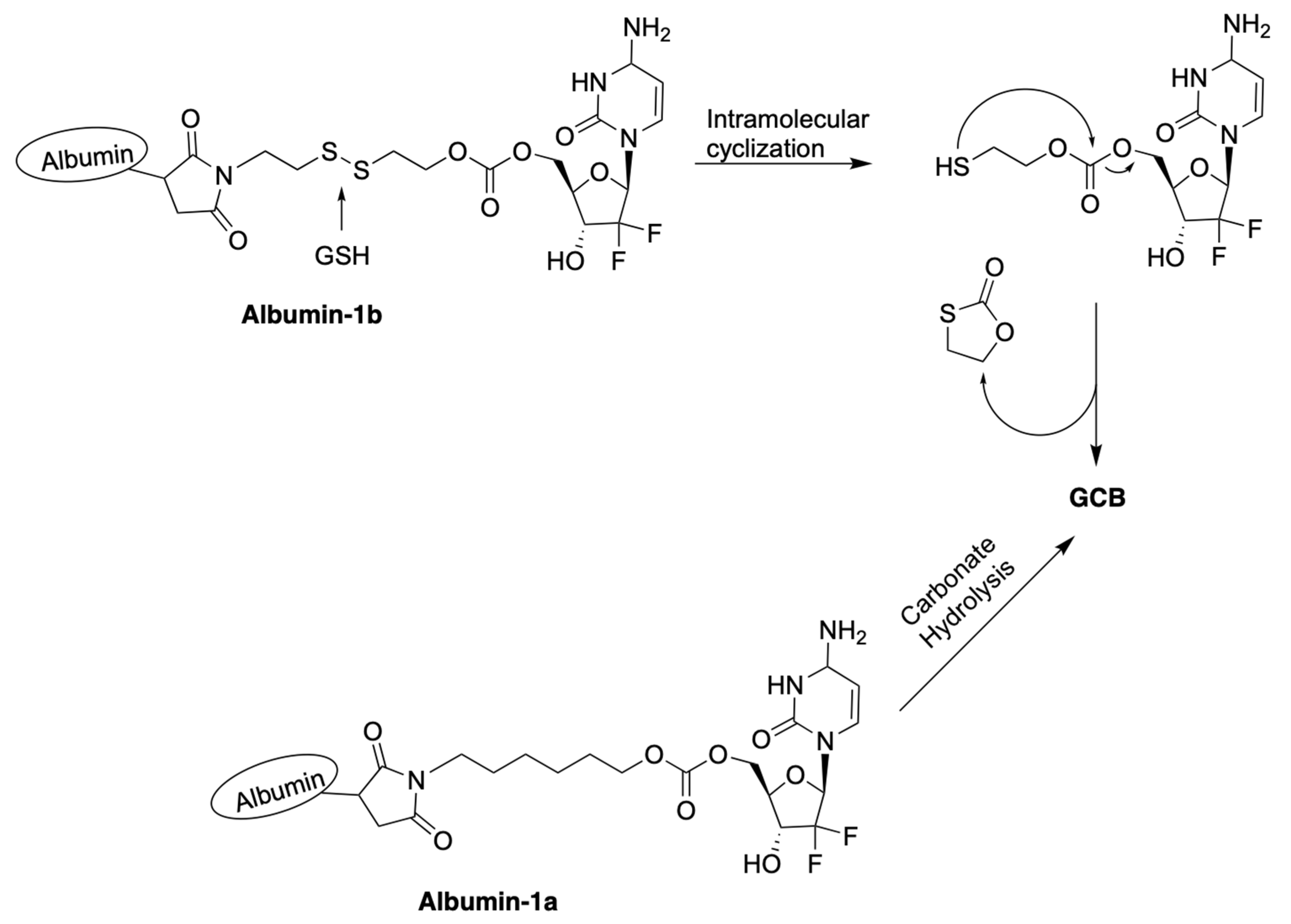
Zhang et al. reported macromolecular constructs containing albumin-conjugated prodrugs of GCB, termed Albumin-1a and Albumin-1b. The reported compounds contain albumin conjugated to 5′-OH of GCB via a carbonate linker [26]. Analogous prodrugs with albumin conjugated to the N4-position of the cytosine nucleobase via a carbamate linker were also described. GCB has a relatively short half-life in vivo. Within 9 min in the human body, it undergoes deamination of the cytosine nucleobase by cytidine deaminase (CDA), which results in the formation of the inactive form of GCB, 2′-deoxy-2′,2′-difluorouridine (dFdU) [27]. To overcome this limitation and enhance its efficacy, human serum albumin (HSA) was used as a macromolecular drug carrier. HSA remains in the blood plasma for days after administration. The reported prodrugs can be activated by the following two different mechanisms, as illustrated in Figure 5: (1) hydrolysis of the labile carbonate linkage at 5′-OH of the sugar moiety; (2) reduction of the disulfide linkage by GSH, followed by intramolecular cyclization.
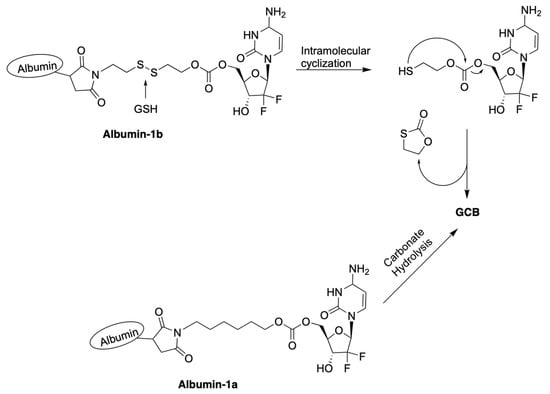
Figure 5.
Proposed mechanisms of activation of prodrugs Albumin-1a and Albumin-1b.
The studies of albumin binding to prodrugs were carried out with bovine serum albumin (BSA). All of the reported prodrugs attached to BSA very fast. Within 1 min of incubation, over 90% of the prodrugs had coupled to BSA. Complete binding was achieved after 30 min of incubation. There was no significant difference between the circular dichroism (CD) data for BSA before and after conjugation, suggesting that the introduction of the prodrug to BSA did not cause significant changes to its secondary structure.
The authors used dithiothreitol (DTT) to study the reductive mechanism of prodrug activation. Albumin-1b was treated with 1 μM DTT for 8 h in PBS (pH = 7.4). This treatment resulted in 33% release of the active form of GCB. A high concentration of DTT (1 mM) drastically improved the release to 100%. The stability of the prodrugs against deamination by CDA was investigated in rat blood plasma. The prodrugs and GCB were incubated at 37 °C for 12 h at a concentration of 16 μg/mL. It was observed that Albumin-1b was mostly transformed to active GCB and formed three-times less dFdU than GCB alone. The other three prodrugs produced no more than 380 ng/mL of dFdU per hour. These results strongly suggest that the prodrugs are less prone to degradation by CDA than GCB.
Antitumor efficacy was investigated in mice bearing 4T1 xenografts. The prodrugs and GCB were administered in five injections of an equivalent dose of 8 mg/kg GCB. Unexpectedly, the prodrug Albumin-1a, which contains a carbonate linkage bond and an all-carbon linker lacking a disulfide bond, was found to be the most potent anticancer agent. Treatment with Albumin-1b resulted in a reduction in the tumor volume by two-thirds. The authors attributed this result to several factors, as follows: (1) improved pharmacokinetic properties; (2) decreased prodrug deactivation by CDA; (3) enhanced prodrug accumulation in the tumor; (4) improved cellular uptake that is less dependent on nucleic acid transporters; (5) in vivo prodrug activation via cleavage of the carbonate bond.
2.6. Prodrugs That Are Based on Bio-Orthogonal Chemistry
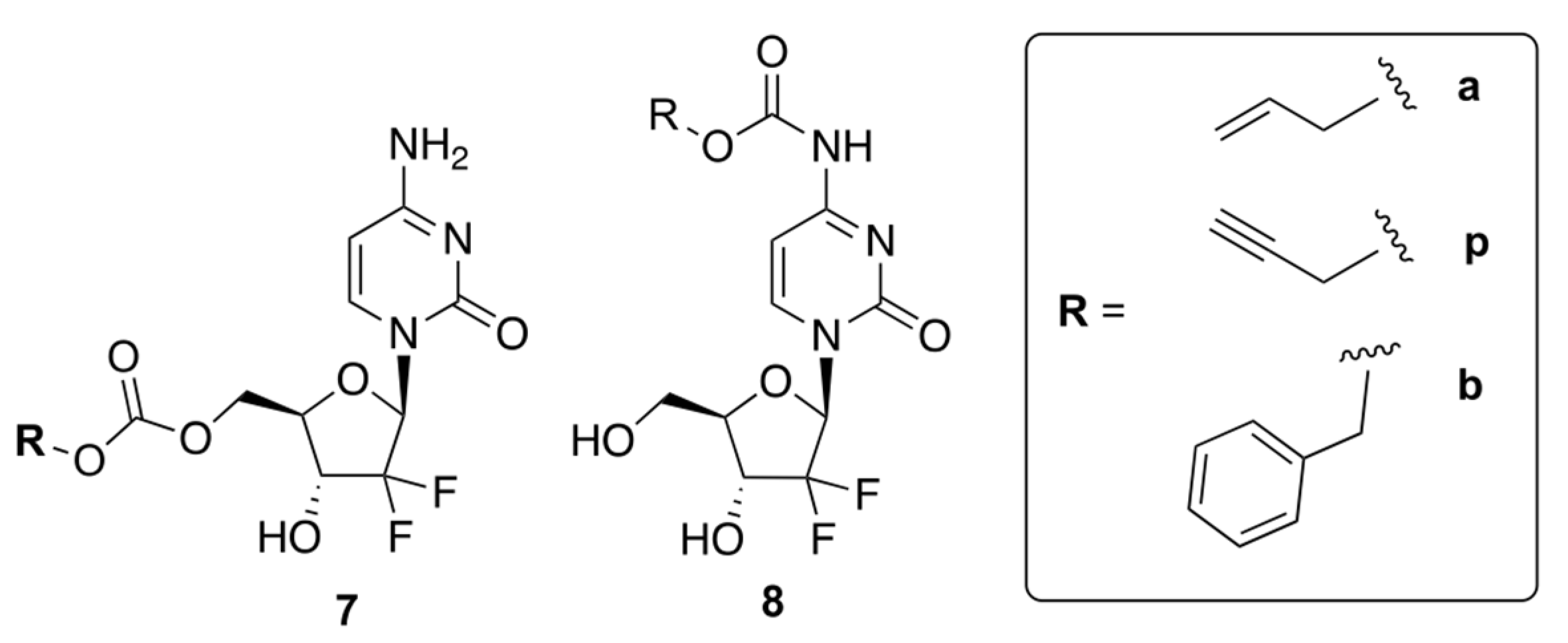
Weiss et al. reported prodrugs of GCB, 7 and 8, which can be uncaged by bio-orthogonal palladium Pd0 chemistry [28]. Transition metal-assisted bio-orthogonal chemistry could be helpful for the controlled release of the drug, which will enhance its efficacy. The authors reported six compounds containing allyl (Alloc), propargyloxycarbonyl (Poc), and carboxybenzyl (Cbz) groups attached to GCB via either a carbamate or carbonate linker (Figure 6). The authors proposed that protecting the 5′-OH would block the formation of cytotoxic metabolites, while masking the N4-position of cytidine would prevent deactivation by CDA.
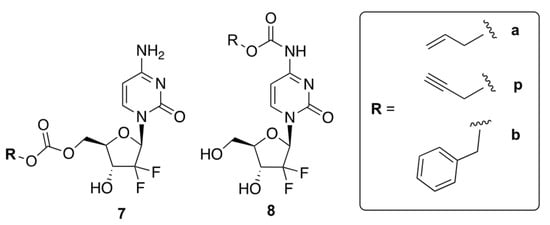
Figure 6.
Prodrugs of GCB that can be uncaged by bio-orthogonal palladium Pd0 chemistry.
The authors carried out cytotoxicity studies using two human cancer cell lines, BxPC3 and Mia PaCa-2. Prodrugs 8 a, p, and b were found to be less cytotoxic than GCB, indicating that the carbamate linker has a shielding effect. On the other hand, prodrugs 7 a, p, and b exhibited similar cytotoxicity to the parent drug. These results indicated that the carbonate linker is cleaved inside the cells, leading to non-specific prodrug activation. Therefore, only compounds 8 a, p, and b were used for further investigation.
The authors carried out HPLC analysis to investigate the release of active GCB from prodrugs 8 a, p, and b. Pd0-functionalized resins were prepared by trapping Pd0 nanoparticles in an amino-functionalized polystyrene matrix. To monitor the effectiveness of Pd0 catalysis, all carbamates 8 a, p, and b (100 μM) were incubated with Pd0 resins (1 mg/mL) dispersed in PBS (300 mOsm/kg, pH = 7.4) at 37 °C for 24 h. As per the analysis of retention times, compound 8 b only generated negligible amounts of active GCB, while 8 a and 8 p produced notable amounts of active GCB. Compound 8 p showed fast and robust unmasking of the carbamate group, with a half-life of less than 6 h. This was consistent with the previously reported palladium-driven catalysis of propargyl groups [29].
The Pd0-catalyzed bio-orthogonal in situ generation of active GCB from the prodrugs 8 a, p, and b was further studied in BxPC-3 and Mia PaCa-2 cell lines. The cells were treated with the prodrugs and Pd0 resins, and the observed cytotoxicities were compared to GCB. As anticipated, prodrugs 8 a and 8 p showed the strongest effect, while prodrug 8 b was not activated by Pd0. Among all the tested compounds, 8 p had the highest potential to generate active GCB in the Pd0-catalyzed bio-orthogonal reaction. In conclusion, the authors emphasized that the bio-orthogonal reaction between the carbamate prodrugs and Pd0 has great potential as a targeted cancer therapy.
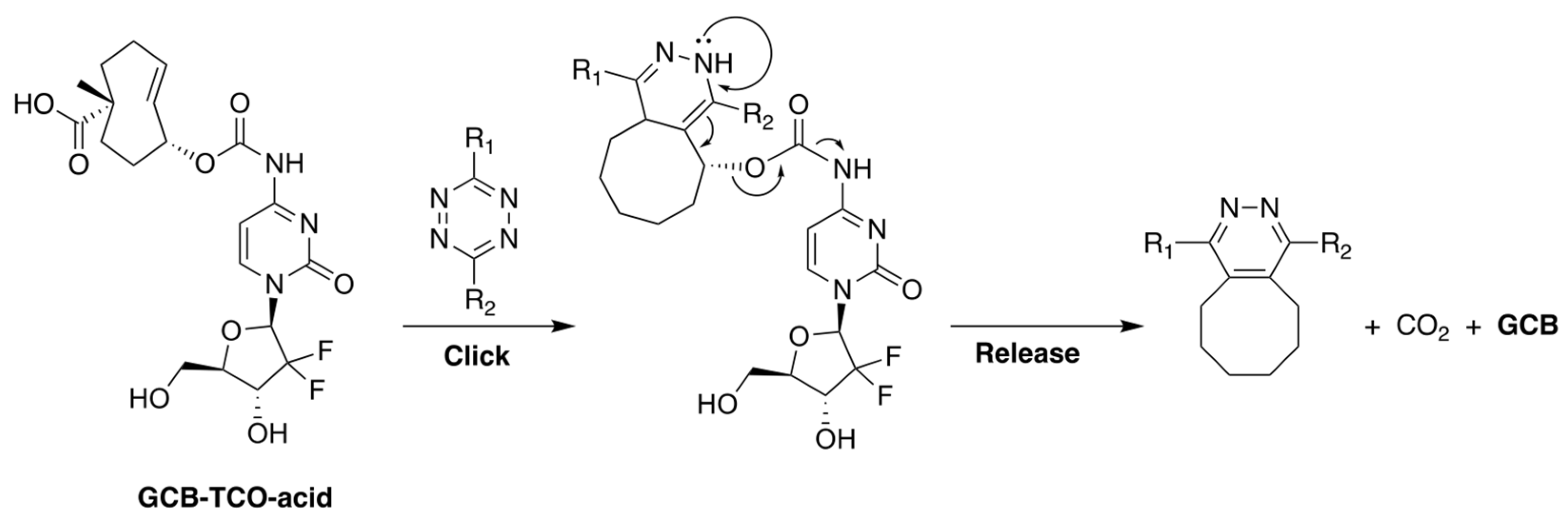
Our group, in collaboration with Shasqi, Inc., recently reported a prodrug of GCB, shown in Figure 7, whose activation is triggered by bio-orthogonal inverse electron demand Diels-Alder (IEDDA) chemistry between trans-cyclooctene (TCO) and tetrazine (Tz) [30]. The prodrug, termed GCB-TCO-acid, contains TCO attached to the N4-position of the nucleobase via a carbamate bond. In the caged form, the prodrug is shielded from deamination by CDA. A carboxylic acid moiety was installed on TCO to improve the prodrug’s aqueous solubility. Upon reaction with Tz, the prodrug is expected to be activated via the bond-cleaving bio-orthogonal chemistry mechanism described in Figure 7 [31].
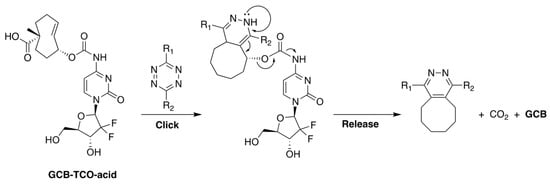
Figure 7.
The structure of GCB-TCO-acid and the mechanism of its activation by Tz via bond-cleaving bio-orthogonal IEDDA chemistry.
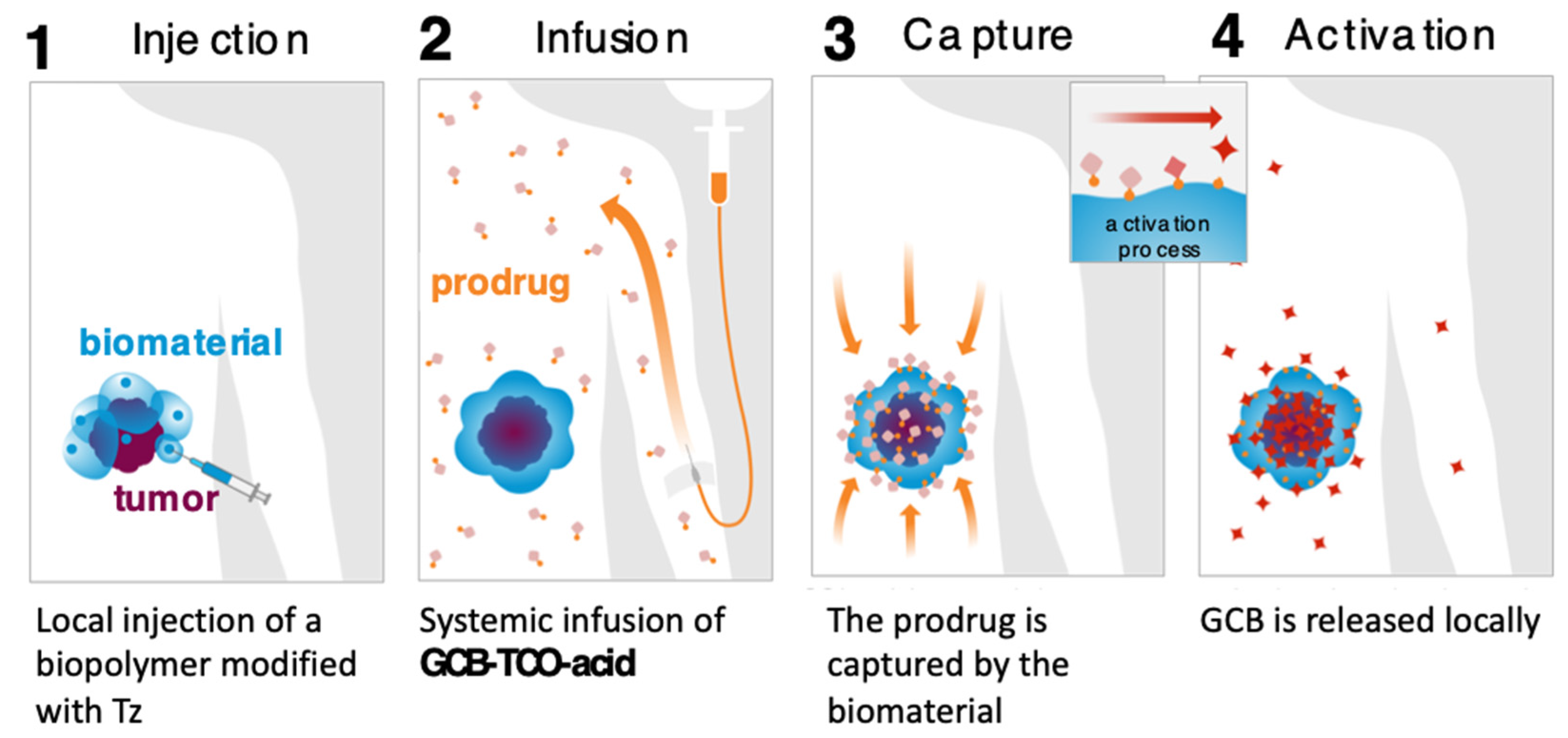
We carried out an assessment of the prodrug’s in vitro properties to determine that it is stable in mouse blood plasma and has a solubility of 3 mg/mL in PBS. The MTT studies with MC38 cells (murine colon adenocarcinoma) showed that the prodrug was 8.7-times less cytotoxic than GCB (IC50 = 3 nM for GCB; IC50 = 26 nM for GCB-TCO-acid). We envisioned that GCB-TCO-acid could be used in conjunction with the CAPAC platform, as shown in Figure 8. The biomaterial-based Click Activated Protodrugs Against Cancer (CAPAC) platform consists of a tetrazine-modified sodium hyaluronate-based biopolymer, SQL70, injected near the tumor site, followed by systemic doses of GCB-TCO-acid. The prodrug is captured locally by the biopolymer through an IEDDA reaction, followed by conversion to active GCB at the tumor site.
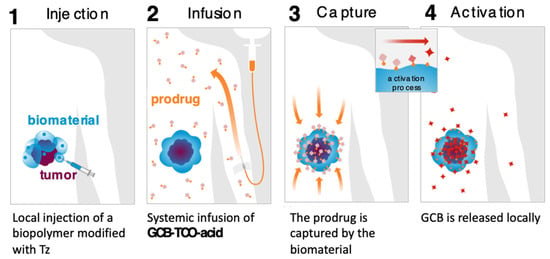
Figure 8.
CAPAC platform for local activation of systemically administered prodrugs to treat cancer.
We evaluated the antitumor effects of GCB-TCO-acid in combination with the reported Tz-functionalized biopolymer SQL70 [32]. This study was carried out in immune-competent mice bearing MC38 xenografts. GCB-TCO-acid was administered IV at a dose of 55 mg/kg, once daily, for five consecutive days. The prodrug treatment correlated with the anti-tumor efficacy (unpublished data). However, because of inadequate inactivation, the administration of high doses of GCB-TCO-acid was associated with severe neurotoxicity. Further work will have to be conducted in the future to design a prodrug of GCB with a higher level of inactivation that can be tolerated more safely at high doses.
3. Conclusions
In conclusion, this review described a number of different prodrug strategies aimed at addressing the shortcomings of FDA-approved GCB-based chemotherapy, such as enzymatic deamination, fast systemic clearance, and chemoresistance by downregulation of cellular uptake. The described strategies included GCB modification with enzyme-labile groups, groups that are cleavable by tumor-specific environmental factors, such as ROS, as well as macromolecular approaches and drug delivery approaches. The modification of GCB at the 5′-OH position or N4-position of the cytosine nucleobase can lead to significant deactivation, as was shown by the in vitro MTT studies. The reported carbamate prodrugs tend to be more stable under simulated physiological conditions, while the reported carbonate prodrugs are less stable. However, they presented alternative in vivo activation strategies that led to promising in vivo data. Several of the described reports presented in vivo efficacy data showing that prodrugs of GCB have fewer side effects and can achieve greater anti-tumor efficacy. The reported data offer significant promise that more effective GCB-based anticancer therapies will be developed in the future.
Author Contributions
Writing—original draft preparation, B.P.; writing—review and editing, M.R.; supervision, M.R.; funding acquisition, M.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Institutes of Health grant 1R21CA228997 to M.R.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors acknowledge the staff and facilities of the Life Sciences Research Building at University at Albany for their support.
Conflicts of Interest
Maksim Royzen is an advisor and shareholder of Shasqi, Inc.
References
- Heinemann, V. Gemcitabine in metastatic breast cancer. Expert Rev. Anticancer Ther. 2005, 5, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Hertel, L.W.; Boder, G.B.; Kroin, J.S.; Rinzel, S.M.; Poore, G.A.; Todd, G.C.; Grindey, G.B. Evaluation of the antitumor activity of gemcitabine (2′,2′-difluoro-2′-deoxycytidine). Cancer Res. 1990, 50, 4417. [Google Scholar]
- Beumer, J.H.; Eiseman, J.L.; Parise, R.A.; Joseph, E.; Covey, J.M.; Egorin, M.J. Modulation of gemcitabine (2′,2′-difluoro-2′-deoxycytidine) pharmacokinetics, metabolism, and bioavailability in mice by 3,4,5,6-tetrahydrouridine. Clin. Cancer Res. 2008, 14, 3529–3535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackey, J.R.; Mani, R.S.; Selner, M.; Mowles, D.; Young, J.D.; Belt, J.A.; Crawford, C.R.; Cass, C.E. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998, 58, 4349–4357. [Google Scholar] [PubMed]
- Qi, H.; Lu, J.; Li, J.; Wang, M.; Xu, Y.; Wang, Y.; Zhang, H. Enhanced antitumor activity of monophosphate ester prodrugs of gemcitabine: In vitro and in vivo evaluation. J. Pharm. Sci. 2016, 105, 2966–2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spratlin, J.; Sangha, R.; Glubrecht, D.; Dabbagh, L.; Young, J.D.; Dumontet, C.; Cass, C.; Lai, R.; Mackey, J.R. The absence of human equilibrative nucleoside transporter 1 is associated with reduced survival in patients with gemcitabine-treated pancreas adenocarcinoma. Clin. Cancer Res. 2004, 10, 6956–6961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Hou, Y.; Meng, X.; Ge, C.; Ma, H.; Li, J.; Fang, J. Selective activation of a prodrug by thioredoxin reductase providing a strategy to target cancer cells. Angew. Chem. 2018, 130, 6249–6253. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Han, X.; Liu, R.; Fang, J. Targeting the thioredoxin system for cancer therapy. Trends Pharmacol. Sci. 2017, 38, 794–808. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, K.; Okuda, T.; Mori, S.; Konno, M.; Eguchi, H.; Asai, A.; Koseki, J.; Iwagami, Y.; Yamada, D.; Akita, H.; et al. A hydrogen peroxide activatable gemcitabine prodrug for the selective treatment of pancreatic ductal adenocarcinoma. ChemMedChem 2019, 14, 1384–1391. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.D.; Sun, C.; Lambeth, J.D.; Marshall, F.; Amin, M.; Chung, L.; Petros, J.A.; Arnold, R.S. Increased Nox1 and hydrogen peroxide in prostate cancer. Prostate 2005, 62, 200–207. [Google Scholar] [CrossRef]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Zieba, M.; Suwalski, M.; Kwiatkowska, S.; Piaseka, G.; Grzelewska–Rzymowska, I.; Stolarek, R.; Nowak, D. Comparison of hydrogen peroxide generation and the content of lipid peroxidation products in lung cancer tissue and pulmonary parenchyma. Resp. Med. 2000, 94, 800–805. [Google Scholar] [CrossRef] [Green Version]
- Rice, M.E. H2O2: A dynamic neuromodulator. Neuroscientist 2011, 17, 389–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veal, E.; Day, A. Hydrogen peroxide as a signaling molecule. Antioxid. Redox Signal. 2011, 15, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.R.; Cotter, T.G. Hydrogen peroxide: A Jekyll and Hyde signalling molecule. Cell Death Dis. 2011, 2, e213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef]
- Lippert, A.R.; Van de Bittner, G.C.; Chang, C.J. Boronate oxidation as a bioorthogonal reaction approach for studying the chemistry of hydrogen peroxide in living systems. Accounts Chem. Res. 2011, 44, 793–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Fang, Z.; Jung, H.-Y.; Yoon, J.-H.; Hong, S.-S.; Maeng, H.-J. Synthesis of gemcitabine-threonine amide prodrug effective on pancreatic cancer cells with improved pharmacokinetic properties. Molecules 2018, 23, 2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, E.-Y.; Shim, W.-S.; Chang, J.-E.; Chong, S.; Kim, D.-D.; Chung, S.-J.; Shim, C.-K. Enhanced intracellular accumulation of a non-nucleoside anti-cancer agent via increased uptake of its valine ester prodrug through amino acid transporters. Xenobiotica 2012, 42, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Maeng, H.-J.; Kim, E.-S.; Chough, C.; Joung, M.; Lim, J.W.; Shim, C.-K.; Shim, W.-S. Addition of amino acid moieties to lapatinib increases the anti-cancer effect via amino acid transporters. Biopharm. Drug Dispos. 2014, 35, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Park, J.-H.; Park, S.; Lee, S.Y.; Cho, K.H.; Kim, D.-D.; Shim, W.-S.; Yoon, I.-S.; Cho, H.-J.; Maeng, H.-J. Enhanced cellular uptake and pharmacokinetic characteristics of doxorubicin-valine amide prodrug. Molecules 2016, 21, 1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.-E.; Jin, H.-E.; Hong, S.-S. Targeting L-type amino acid transporter 1 for anticancer therapy: Clinical impact from diagnostics to therapeutics. Expert Opin. Ther. Targets 2015, 19, 1319–1337. [Google Scholar] [CrossRef] [PubMed]
- Kaira, K.; Sunose, Y.; Arakawa, K.; Ogawa, T.; Sunaga, N.; Shimizu, K.; Tominaga, H.; Oriuchi, N.; Itoh, H.; Nagamori, S.; et al. Prognostic significance of L-type amino-acid transporter 1 expression in surgically resected pancreatic cancer. Brit. J. Cancer 2012, 107, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Ichinoe, M.; Mikami, T.; Yoshida, T.; Igawa, I.; Tsuruta, T.; Nakada, N.; Anzai, N.; Suzuki, Y.; Endou, H.; Okayasu, I. High expression of L-type amino-acid transporter 1 (LAT1) in gastric carcinomas: Comparison with non-cancerous lesions. Pathol. Int. 2011, 61, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, K.; Na, K.; Li, D.; Li, Z.; Zhao, D.; Zhong, L.; Wang, M.; Kou, L.; Luo, C.; et al. Striking a balance between carbonate/carbamate linkage bond- and reduction-sensitive disulfide bond-bearing linker for tailored controlled release: In situ covalent-albumin-binding gemcitabine prodrugs promote bioavailability and tumor accumulation. J. Med. Chem. 2018, 61, 4904–4917. [Google Scholar] [CrossRef]
- Slusarczyk, M.; Lopez, M.H.; Balzarini, J.; Mason, M.; Jiang, W.G.; Blagden, S.; Thompson, E.; Ghazaly, E.; McGuigan, C. Application of ProTide technology to gemcitabine: A successful approach to overcome the key cancer Resistance mechanisms leads to a new agent (NUC-1031) in clinical development. J. Med. Chem. 2014, 57, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.T.; Dawson, J.C.; Fraser, C.; Rybski, W.; Torres-Sánchez, C.; Bradley, M.; Patton, E.E.; Carragher, N.O.; Unciti-Broceta, A. Development and bioorthogonal activation of palladium-labile prodrugs of gemcitabine. J. Med. Chem. 2014, 57, 5395–5404. [Google Scholar] [CrossRef]
- Li, J.; Yu, J.; Zhao, J.; Wang, J.; Zheng, S.; Lin, S.; Chen, L.; Yang, M.; Jia, S.; Zhang, X.; et al. Palladium-triggered deprotection chemistry for protein activation in living cells. Nat. Chem. 2014, 6, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Yee, N.A.; Srinivasan, S.; Mahmoodi, A.; Zakharian, M.; Oneto, J.M.M.; Royzen, M. Click activated protodrugs against cancer increase the therapeutic potential of chemotherapy through local capture and activation. Chem. Sci. 2021, 12, 1259–1271. [Google Scholar] [CrossRef]
- Versteegen, R.M.; ten Hoeve, W.; Rossin, R.; de Geus, M.A.R.; Janssen, H.M.; Robillard, M.S. Click-to-release from trans-cyclooctenes: Mechanistic insights and expansion of scope from established carbamate to remarkable ether cleavage. Angew. Chem. Int. Ed. 2018, 57, 10494–10499. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Yee, N.A.; Wu, K.; Zakharian, M.; Mahmoodi, A.; Royzen, M.; Mejía Oneto, J.M. SQ3370 activates cytotoxic drug via click chemistry at tumor and elicits sustained responses in injected and non-injected lesions. Adv. Ther. 2021, 4, 2000243. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).