
G-Quadruplex Matters in Tissue-Specific Tumorigenesis by BRCA1 Deficiency


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Tissue-Specific Tumor Susceptibility of BRCA1
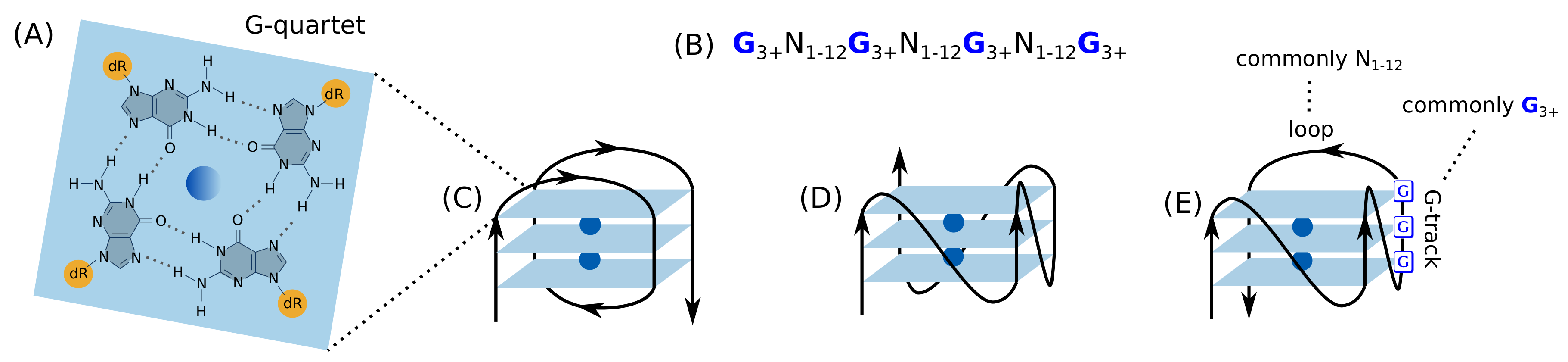
2. G-Quadruplex
2.1. Introduction to the G-Quadruplex
2.2. G4s as the Determinants of Mutagenesis
2.3. G4s Are Key Genomic Structural Elements in Transcriptional Regulation
3. The Role of BRCA1 in Resolving Regulatory G4s That Can Induce DNA Damage
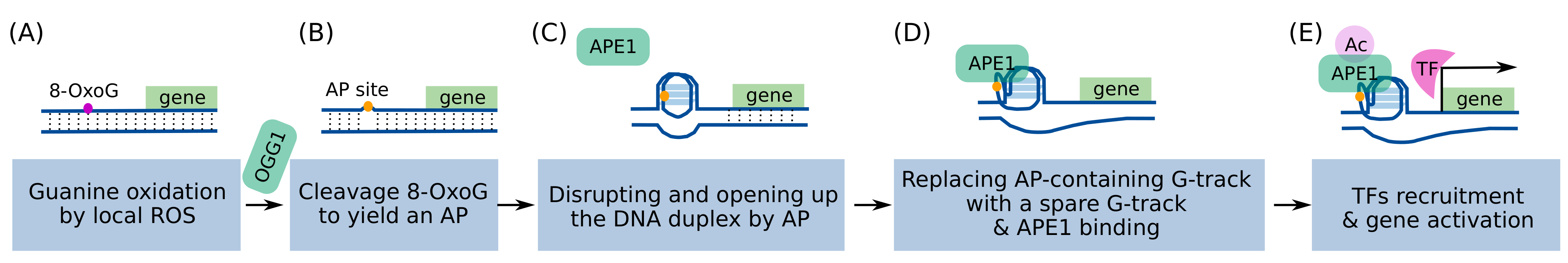
3.1. Increased Levels of Transcriptional Regulatory G4s Can Cause DNA Damage
3.2. The Accumulation of Transcriptional Regulatory G4s and DNA Damage Depends on the BRCA1 Status and This Dependency Is Cell-Type Specific
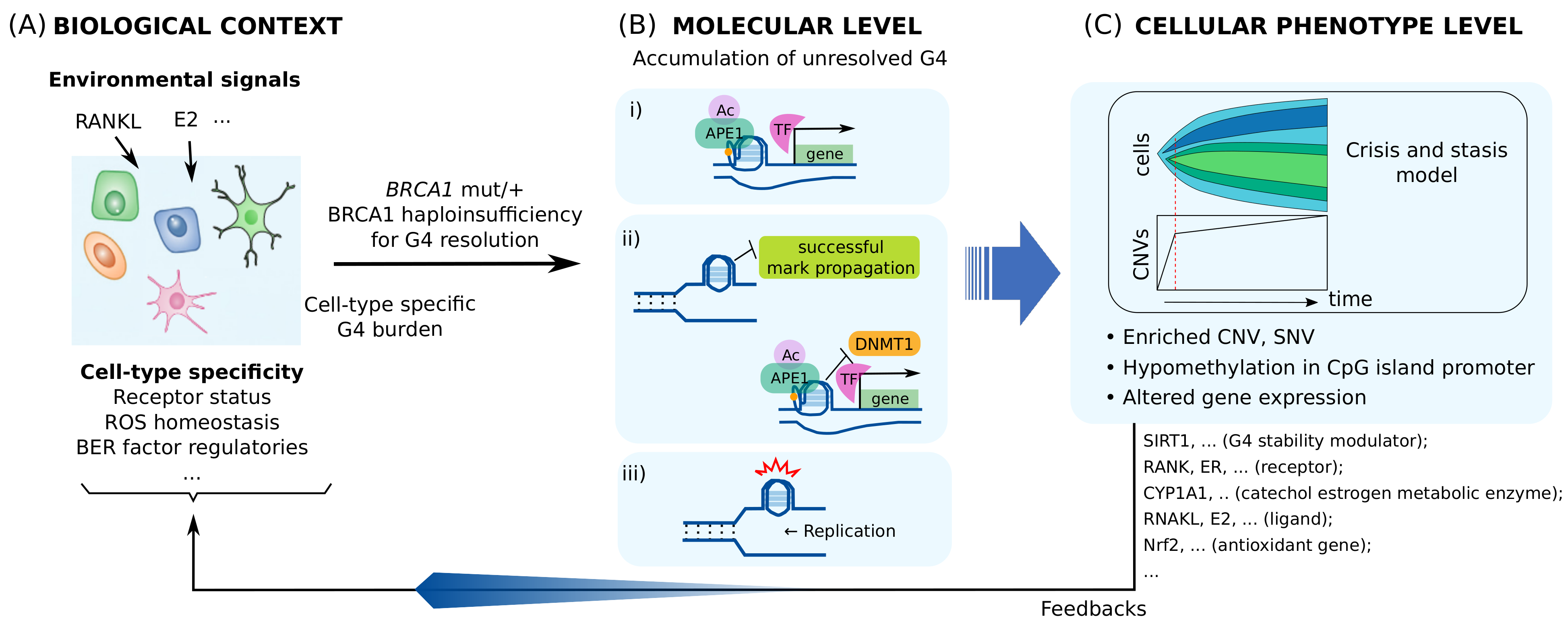
4. Consequence of BRCA1 Haploinsufficiency
4.1. BRCA1 Heterozygosity Cause a Cell-Type Specific Haploinsufficiency for Resolving G4s
4.2. Altered Gene Expression Caused by BRCA1 Haploinsufficiency Can Lead to Cell-Type-Specific Genomic Instability and Premature Senescence
4.3. BRCA1 Insufficiency Causes Multi-Level Heterogeneous Molecular and Cellular Alterations
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A Strong Candidate for the Breast and Ovarian Cancer Susceptibility Gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. How Do Mutations Affecting the Breast Cancer Genes BRCA1 and BRCA2 Cause Cancer Susceptibility? DNA Repair 2019, 81, 102668. [Google Scholar] [CrossRef]
- Ding, L.; Bailey, M.H.; Porta-Pardo, E.; Thorsson, V.; Colaprico, A.; Bertrand, D.; Gibbs, D.L.; Weerasinghe, A.; Huang, K.; Tokheim, C.; et al. Perspective on Oncogenic Processes at the End of the Beginning of Cancer Genomics. Cell 2018, 173, 305–320.e10. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Mooij, T.M.; Roos-Blom, M.-J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of “BRCAness” in Sporadic Cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness Revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive Molecular Portraits of Human Breast Tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform Analysis of 12 Cancer Types Reveals Molecular Classification within and across Tissues of Origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef]
- Jonsson, P.; Bandlamudi, C.; Cheng, M.L.; Srinivasan, P.; Chavan, S.S.; Friedman, N.D.; Rosen, E.Y.; Richards, A.L.; Bouvier, N.; Selcuklu, S.D.; et al. Tumour Lineage Shapes BRCA-Mediated Phenotypes. Nature 2019, 571, 576–579. [Google Scholar] [CrossRef] [PubMed]
- De Talhouet, S.; Peron, J.; Vuilleumier, A.; Friedlaender, A.; Viassolo, V.; Ayme, A.; Bodmer, A.; Treilleux, I.; Lang, N.; Tille, J.-C.; et al. Clinical Outcome of Breast Cancer in Carriers of BRCA1 and BRCA2 Mutations According to Molecular Subtypes. Sci. Rep. 2020, 10, 7073. [Google Scholar] [CrossRef]
- Elledge, S.J.; Amon, A. The BRCA1 Suppressor Hypothesis: An Explanation for the Tissue-Specific Tumor Development in BRCA1 Patients. Cancer Cell 2002, 1, 129–132. [Google Scholar] [CrossRef]
- Schneider, G.; Schmidt-Supprian, M.; Rad, R.; Saur, D. Tissue-Specific Tumorigenesis: Context Matters. Nat. Rev. Cancer 2017, 17, 239–253. [Google Scholar] [CrossRef]
- Chen, C.-C.; Feng, W.; Lim, P.X.; Kass, E.M.; Jasin, M. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu. Rev. Cancer Biol. 2018, 2, 313–336. [Google Scholar] [CrossRef] [PubMed]
- Tye, S.; Ronson, G.E.; Morris, J.R. A Fork in the Road: Where Homologous Recombination and Stalled Replication Fork Protection Part Ways. Semin. Cell Dev. Biol. 2020, 113, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, K.N.; Wubbenhorst, B.; Wenz, B.M.; De Sloover, D.; Pluta, J.; Emery, L.; Barrett, A.; Kraya, A.A.; Anastopoulos, I.N.; Yu, S.; et al. BRCA Locus-Specific Loss of Heterozygosity in Germline BRCA1 and BRCA2 Carriers. Nat. Commun. 2017, 8, 319. [Google Scholar] [CrossRef] [PubMed]
- Haigis, K.M.; Cichowski, K.; Elledge, S.J. Tissue-Specificity in Cancer: The Rule, Not the Exception. Science 2019, 363, 1150–1151. [Google Scholar] [CrossRef]
- Singh, A.K.; Yu, X. Tissue-Specific Carcinogens as Soil to Seed BRCA1/2-Mutant Hereditary Cancers. Trends Cancer 2020, 6, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Liehr, J.G. Is Estradiol a Genotoxic Mutagenic Carcinogen?1. Endocr. Rev. 2000, 21, 40–54. [Google Scholar] [CrossRef]
- Gorrini, C.; Gang, B.P.; Bassi, C.; Wakeham, A.; Baniasadi, S.P.; Hao, Z.; Li, W.Y.; Cescon, D.W.; Li, Y.-T.; Molyneux, S.; et al. Estrogen Controls the Survival of BRCA1-Deficient Cells via a PI3K–NRF2-Regulated Pathway. Proc. Natl. Acad. Sci. USA 2014, 111, 4472–4477. [Google Scholar] [CrossRef]
- Zhang, X.; Li, R. BRCA1-Dependent Transcriptional Regulation: Implication in Tissue-Specific Tumor Suppression. Cancers 2018, 10, 513. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, J.; Tacconi, E.M.C.; Folio, C.; Badie, S.; Porru, M.; Klare, K.; Tumiati, M.; Markkanen, E.; Halder, S.; Ryan, A.; et al. Targeting BRCA1 and BRCA2 Deficiencies with G-Quadruplex-Interacting Compounds. Mol. Cell 2016, 61, 449–460. [Google Scholar] [CrossRef]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Santos, N.D.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 Is a DNA G-Quadruplex Stabilizer with Selective Lethality in BRCA1/2 Deficient Tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef]
- Hu, M.-H.; Wu, T.-Y.; Huang, Q.; Jin, G. New Substituted Quinoxalines Inhibit Triple-Negative Breast Cancer by Specifically Downregulating the c-MYC Transcription. Nucleic Acids Res. 2019, 47, 10529–10542. [Google Scholar] [CrossRef] [PubMed]
- Sanij, E.; Hannan, K.M.; Xuan, J.; Yan, S.; Ahern, J.E.; Trigos, A.S.; Brajanovski, N.; Son, J.; Chan, K.T.; Kondrashova, O.; et al. CX-5461 Activates the DNA Damage Response and Demonstrates Therapeutic Efficacy in High-Grade Serous Ovarian Cancer. Nat. Commun. 2020, 11, 2641. [Google Scholar] [CrossRef]
- Hu, M.-H.; Lin, J.-H. New Dibenzoquinoxalines Inhibit Triple-Negative Breast Cancer Growth by Dual Targeting of Topoisomerase 1 and the c-MYC G-Quadruplex. J. Med. Chem. 2021, 64, 6720–6729. [Google Scholar] [CrossRef]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA Secondary Structures: Stability and Function of G-Quadruplex Structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef]
- Parkinson, G.N.; Lee, M.P.H.; Neidle, S. Crystal Structure of Parallel Quadruplexes from Human Telomeric DNA. Nature 2002, 417, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct Evidence for a G-Quadruplex in a Promoter Region and Its Targeting with a Small Molecule to Repress c-MYC Transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef] [PubMed]
- Phan, A.T.; Kuryavyi, V.; Gaw, H.Y.; Patel, D.J. Small-Molecule Interaction with a Five-Guanine-Tract G-Quadruplex Structure from the Human MYC Promoter. Nat. Chem. Biol. 2005, 1, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, K.M.; Saunders, L.B.; Simmons, J.K.; Leon, E.; Calabrese, D.R.; Zhang, S.; Michalowski, A.; Gareiss, P.; Mock, B.A.; Schneekloth, J.S. Small Molecule Microarrays Enable the Identification of a Selective, Quadruplex-Binding Inhibitor of MYC Expression. ACS Chem. Biol. 2016, 11, 139–148. [Google Scholar] [CrossRef]
- Calabrese, D.R.; Chen, X.; Leon, E.C.; Gaikwad, S.M.; Phyo, Z.; Hewitt, W.M.; Alden, S.; Hilimire, T.A.; He, F.; Michalowski, A.M.; et al. Chemical and Structural Studies Provide a Mechanistic Basis for Recognition of the MYC G-Quadruplex. Nat. Commun. 2018, 9, 4229. [Google Scholar] [CrossRef] [PubMed]
- Todd, A.K.; Johnston, M.; Neidle, S. Highly Prevalent Putative Quadruplex Sequence Motifs in Human DNA. Nucleic Acids Res. 2005, 33, 2901–2907. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.L.; Balasubramanian, S. Prevalence of Quadruplexes in the Human Genome. Nucleic Acids Res. 2005, 33, 2908–2916. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.L.; Balasubramanian, S. G-Quadruplexes in Promoters throughout the Human Genome. Nucleic Acids Res. 2007, 35, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Brooks, T.A.; Kendrick, S.; Hurley, L. Making Sense of G-Quadruplex and i-Motif Functions in Oncogene Promoters. FEBS J. 2010, 277, 3459–3469. [Google Scholar] [CrossRef]
- Chambers, V.S.; Marsico, G.; Boutell, J.M.; Di Antonio, M.; Smith, G.P.; Balasubramanian, S. High-Throughput Sequencing of DNA G-Quadruplex Structures in the Human Genome. Nat. Biotechnol. 2015, 33, 877–881. [Google Scholar] [CrossRef] [PubMed]
- Hänsel-Hertsch, R.; Beraldi, D.; Lensing, S.V.; Marsico, G.; Zyner, K.; Parry, A.; Di Antonio, M.; Pike, J.; Kimura, H.; Narita, M.; et al. G-Quadruplex Structures Mark Human Regulatory Chromatin. Nat. Genet. 2016, 48, 1267–1272. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-Quadruplexes in Gene Promoters: A Novel Anticancer Strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging Landscape of Oncogenic Signatures across Human Cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef]
- Di Antonio, M.; Ponjavic, A.; Radzevičius, A.; Ranasinghe, R.T.; Catalano, M.; Zhang, X.; Shen, J.; Needham, L.-M.; Lee, S.F.; Klenerman, D.; et al. Single-Molecule Visualization of DNA G-Quadruplex Formation in Live Cells. Nat. Chem. 2020, 12, 832–837. [Google Scholar] [CrossRef]
- Neidle, S. Quadruplex Nucleic Acids as Novel Therapeutic Targets. J. Med. Chem. 2016, 59, 5987–6011. [Google Scholar] [CrossRef]
- Neidle, S. Quadruplex Nucleic Acids as Targets for Anticancer Therapeutics. Nat. Rev. Chem. 2017, 1, 1–10. [Google Scholar] [CrossRef]
- Asamitsu, S.; Obata, S.; Yu, Z.; Bando, T.; Sugiyama, H. Recent Progress of Targeted G-Quadruplex-Preferred Ligands Toward Cancer Therapy. Molecules 2019, 24, 429. [Google Scholar] [CrossRef]
- Carvalho, J.; Mergny, J.-L.; Salgado, G.F.; Queiroz, J.A.; Cruz, C. G-Quadruplex, Friend or Foe: The Role of the G-Quartet in Anticancer Strategies. Trends Mol. Med. 2020, 26, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Hänsel-Hertsch, R.; Di Antonio, M.; Balasubramanian, S. DNA G-Quadruplexes in the Human Genome: Detection, Functions and Therapeutic Potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, J.; Adhikari, S.; Balasubramanian, S. The Structure and Function of DNA G-Quadruplexes. Trends Chem. 2020, 2, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Varshney, D.; Spiegel, J.; Zyner, K.; Tannahill, D.; Balasubramanian, S. The Regulation and Functions of DNA and RNA G-Quadruplexes. Nat. Rev. Mol. Cell Biol. 2020, 21, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Bacolla, A.; Jaworski, A.; Larson, J.E.; Jakupciak, J.P.; Chuzhanova, N.; Abeysinghe, S.S.; O’Connell, C.D.; Cooper, D.N.; Wells, R.D. Breakpoints of Gross Deletions Coincide with Non-B DNA Conformations. Proc. Natl. Acad. Sci. USA 2004, 101, 14162–14167. [Google Scholar] [CrossRef]
- Georgakopoulos-Soares, I.; Morganella, S.; Jain, N.; Hemberg, M.; Nik-Zainal, S. Noncanonical Secondary Structures Arising from Non-B DNA Motifs Are Determinants of Mutagenesis. Genome Res. 2018, 28, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Guiblet, W.M.; Cremona, M.A.; Harris, R.S.; Chen, D.; Eckert, K.A.; Chiaromonte, F.; Huang, Y.-F.; Makova, K.D. Non-B DNA: A Major Contributor to Small- and Large-Scale Variation in Nucleotide Substitution Frequencies across the Genome. Nucleic Acids Res. 2021, 49, 1497–1516. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, A.; Sabarinathan, R.; Lopez-Bigas, N. Local Determinants of the Mutational Landscape of the Human Genome. Cell 2019, 177, 101–114. [Google Scholar] [CrossRef]
- Supek, F.; Lehner, B. Scales and Mechanisms of Somatic Mutation Rate Variation across the Human Genome. DNA Repair 2019, 81, 102647. [Google Scholar] [CrossRef]
- Ong, C.-T.; Corces, V.G. CTCF: An Architectural Protein Bridging Genome Topology and Function. Nat. Rev. Genet. 2014, 15, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, A.S.; Li, C.H.; Zamudio, A.V.; Sigova, A.A.; Hannett, N.M.; Day, D.S.; Abraham, B.J.; Cohen, M.A.; Nabet, B.; Buckley, D.L.; et al. YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 2017, 171, 1573–1588.e28. [Google Scholar] [CrossRef] [PubMed]
- Rowley, M.J.; Corces, V.G. Organizational Principles of 3D Genome Architecture. Nat. Rev. Genet. 2018, 19, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Jin, W.; Cui, K.; Rodrigez, J.; Hu, G.; Zhang, Z.; Larson, D.R.; Zhao, K. CTCF-Mediated Enhancer-Promoter Interaction Is a Critical Regulator of Cell-to-Cell Variation of Gene Expression. Mol. Cell 2017, 67, 1049–1058.e6. [Google Scholar] [CrossRef]
- Hanssen, L.L.P.; Kassouf, M.T.; Oudelaar, A.M.; Biggs, D.; Preece, C.; Downes, D.J.; Gosden, M.; Sharpe, J.A.; Sloane-Stanley, J.A.; Hughes, J.R.; et al. Tissue-Specific CTCF–Cohesin-Mediated Chromatin Architecture Delimits Enhancer Interactions and Function in Vivo. Nat. Cell Biol. 2017, 19, 952–961. [Google Scholar] [CrossRef]
- Schoenfelder, S.; Fraser, P. Long-Range Enhancer–Promoter Contacts in Gene Expression Control. Nat. Rev. Genet. 2019, 20, 437–455. [Google Scholar] [CrossRef]
- Schuijers, J.; Manteiga, J.C.; Weintraub, A.S.; Day, D.S.; Zamudio, A.V.; Hnisz, D.; Lee, T.I.; Young, R.A. Transcriptional Dysregulation of MYC Reveals Common Enhancer-Docking Mechanism. Cell Rep. 2018, 23, 349–360. [Google Scholar] [CrossRef]
- Kubo, N.; Ishii, H.; Xiong, X.; Bianco, S.; Meitinger, F.; Hu, R.; Hocker, J.D.; Conte, M.; Gorkin, D.; Yu, M.; et al. Promoter-Proximal CTCF Binding Promotes Distal Enhancer-Dependent Gene Activation. Nat. Struct. Mol. Biol. 2021, 28, 152–161. [Google Scholar] [CrossRef]
- Schuster-Böckler, B.; Lehner, B. Chromatin Organization Is a Major Influence on Regional Mutation Rates in Human Cancer Cells. Nature 2012, 488, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Katainen, R.; Dave, K.; Pitkänen, E.; Palin, K.; Kivioja, T.; Välimäki, N.; Gylfe, A.E.; Ristolainen, H.; Hänninen, U.A.; Cajuso, T.; et al. CTCF/Cohesin-Binding Sites Are Frequently Mutated in Cancer. Nat. Genet. 2015, 47, 818–821. [Google Scholar] [CrossRef]
- Kaiser, V.B.; Taylor, M.S.; Semple, C.A. Mutational Biases Drive Elevated Rates of Substitution at Regulatory Sites across Cancer Types. PLoS Genet. 2016, 12, e1006207. [Google Scholar] [CrossRef] [PubMed]
- Poulos, R.C.; Thoms, J.A.I.; Guan, Y.F.; Unnikrishnan, A.; Pimanda, J.E.; Wong, J.W.H. Functional Mutations Form at CTCF-Cohesin Binding Sites in Melanoma Due to Uneven Nucleotide Excision Repair across the Motif. Cell Rep. 2016, 17, 2865–2872. [Google Scholar] [CrossRef]
- Guo, Y.A.; Chang, M.M.; Huang, W.; Ooi, W.F.; Xing, M.; Tan, P.; Skanderup, A.J. Mutation Hotspots at CTCF Binding Sites Coupled to Chromosomal Instability in Gastrointestinal Cancers. Nat. Commun. 2018, 9, 1520. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.M.; Martinez-Fundichely, A.; Diaz, B.J.; Aronson, B.; Cuykendall, T.; MacKay, M.; Dhingra, P.; Wong, E.W.P.; Chi, P.; Apostolou, E.; et al. Identification of Cancer Drivers at CTCF Insulators in 1962 Whole Genomes. Cell Syst. 2019, 8, 446–455.e8. [Google Scholar] [CrossRef]
- Hou, Y.; Li, F.; Zhang, R.; Li, S.; Liu, H.; Qin, Z.S.; Sun, X. Integrative Characterization of G-Quadruplexes in the Three-Dimensional Chromatin Structure. Epigenetics 2019, 14, 894–911. [Google Scholar] [CrossRef]
- Tikhonova, P.; Pavlova, I.; Isaakova, E.; Tsvetkov, V.; Bogomazova, A.; Vedekhina, T.; Luzhin, A.V.; Sultanov, R.; Severov, V.; Klimina, K.; et al. DNA G-Quadruplexes Contribute to CTCF Recruitment. Int. J. Mol. Sci. 2021, 22, 7090. [Google Scholar] [CrossRef]
- Li, L.; Williams, P.; Ren, W.; Wang, M.Y.; Gao, Z.; Miao, W.; Huang, M.; Song, J.; Wang, Y. YY1 Interacts with Guanine Quadruplexes to Regulate DNA Looping and Gene Expression. Nat. Chem. Biol. 2021, 17, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Stamatoyannopoulos, J.A.; Adzhubei, I.; Thurman, R.E.; Kryukov, G.V.; Mirkin, S.M.; Sunyaev, S.R. Human Mutation Rate Associated with DNA Replication Timing. Nat. Genet. 2009, 41, 393–395. [Google Scholar] [CrossRef]
- Sugimoto, N.; Maehara, K.; Yoshida, K.; Ohkawa, Y.; Fujita, M. Genome-Wide Analysis of the Spatiotemporal Regulation of Firing and Dormant Replication Origins in Human Cells. Nucleic Acids Res. 2018, 46, 6683–6696. [Google Scholar] [CrossRef] [PubMed]
- Prorok, P.; Artufel, M.; Aze, A.; Coulombe, P.; Peiffer, I.; Lacroix, L.; Guédin, A.; Mergny, J.-L.; Damaschke, J.; Schepers, A.; et al. Involvement of G-Quadruplex Regions in Mammalian Replication Origin Activity. Nat. Commun. 2019, 10, 3274. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; De, S.; Michor, F. DNA Replication Timing and Higher-Order Nuclear Organization Determine Single-Nucleotide Substitution Patterns in Cancer Genomes. Nat. Commun. 2013, 4, 1502. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Karlić, R.; Koren, A.; Thurman, R.; Sandstrom, R.; Lawrence, M.S.; Reynolds, A.; Rynes, E.; Vlahoviček, K.; Stamatoyannopoulos, J.A.; et al. Cell-of-Origin Chromatin Organization Shapes the Mutational Landscape of Cancer. Nature 2015, 518, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Marchal, C.; Sima, J.; Gilbert, D.M. Control of DNA Replication Timing in the 3D Genome. Nat. Rev. Mol. Cell Biol. 2019, 1–17. [Google Scholar] [CrossRef]
- Su, Q.P.; Zhao, Z.W.; Meng, L.; Ding, M.; Zhang, W.; Li, Y.; Liu, M.; Li, R.; Gao, Y.-Q.; Xie, X.S.; et al. Superresolution Imaging Reveals Spatiotemporal Propagation of Human Replication Foci Mediated by CTCF-Organized Chromatin Structures. Proc. Natl. Acad. Sci. USA 2020, 117, 15036–15046. [Google Scholar] [CrossRef]
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell 2019, 73, 398–411. [Google Scholar] [CrossRef]
- García-Muse, T.; Aguilera, A. R Loops: From Physiological to Pathological Roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef]
- Niehrs, C.; Luke, B. Regulatory R-Loops as Facilitators of Gene Expression and Genome Stability. Nat. Rev. Mol. Cell Biol. 2020, 21, 167–178. [Google Scholar] [CrossRef]
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chédin, F. R-Loop Formation Is a Distinctive Characteristic of Unmethylated Human CpG Island Promoters. Mol. Cell 2012, 45, 814–825. [Google Scholar] [CrossRef]
- Ginno, P.A.; Lim, Y.W.; Lott, P.L.; Korf, I.; Chédin, F. GC Skew at the 5′ and 3′ Ends of Human Genes Links R-Loop Formation to Epigenetic Regulation and Transcription Termination. Genome Res. 2013, 23, 1590–1600. [Google Scholar] [CrossRef]
- Sanz, L.A.; Hartono, S.R.; Lim, Y.W.; Steyaert, S.; Rajpurkar, A.; Ginno, P.A.; Xu, X.; Chédin, F. Prevalent, Dynamic, and Conserved R-Loop Structures Associate with Specific Epigenomic Signatures in Mammals. Mol. Cell 2016, 63, 167–178. [Google Scholar] [CrossRef]
- Kuznetsov, V.A.; Bondarenko, V.; Wongsurawat, T.; Yenamandra, S.P.; Jenjaroenpun, P. Toward Predictive R-Loop Computational Biology: Genome-Scale Prediction of R-Loops Reveals Their Association with Complex Promoter Structures, G-Quadruplexes and Transcriptionally Active Enhancers. Nucleic Acids Res. 2018, 46, 7566–7585. [Google Scholar] [CrossRef]
- Lim, G.; Hohng, S. Single-Molecule Fluorescence Studies on Cotranscriptional G-Quadruplex Formation Coupled with R-Loop Formation. Nucleic Acids Res. 2020, 48, 9195–9203. [Google Scholar] [CrossRef]
- Lee, C.-Y.; McNerney, C.; Ma, K.; Zhao, W.; Wang, A.; Myong, S. R-Loop Induced G-Quadruplex in Non-Template Promotes Transcription by Successive R-Loop Formation. Nat. Commun. 2020, 11, 3392. [Google Scholar] [CrossRef]
- Duquette, M.L.; Handa, P.; Vincent, J.A.; Taylor, A.F.; Maizels, N. Intracellular Transcription of G-Rich DNAs Induces Formation of G-Loops, Novel Structures Containing G4 DNA. Genes Dev. 2004, 18, 1618–1629. [Google Scholar] [CrossRef]
- Shen, J.; Varshney, D.; Simeone, A.; Zhang, X.; Adhikari, S.; Tannahill, D.; Balasubramanian, S. Promoter G-Quadruplex Folding Precedes Transcription and Is Controlled by Chromatin. Genome Biol. 2021, 22, 143. [Google Scholar] [CrossRef]
- De Magis, A.; Manzo, S.G.; Russo, M.; Marinello, J.; Morigi, R.; Sordet, O.; Capranico, G. DNA Damage and Genome Instability by G-Quadruplex Ligands Are Mediated by R Loops in Human Cancer Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Miglietta, G.; Russo, M.; Capranico, G. G-Quadruplex–R-Loop Interactions and the Mechanism of Anticancer G-Quadruplex Binders. Nucleic Acids Res. 2020, 48, 11942–11957. [Google Scholar] [CrossRef] [PubMed]
- Cogoi, S.; Shchekotikhin, A.E.; Xodo, L.E. HRAS Is Silenced by Two Neighboring G-Quadruplexes and Activated by MAZ, a Zinc-Finger Transcription Factor with DNA Unfolding Property. Nucleic Acids Res. 2014, 42, 8379–8388. [Google Scholar] [CrossRef] [PubMed]
- Kouzine, F.; Wojtowicz, D.; Baranello, L.; Yamane, A.; Nelson, S.; Resch, W.; Kieffer-Kwon, K.-R.; Benham, C.J.; Casellas, R.; Przytycka, T.M.; et al. Permanganate/S1 Nuclease Footprinting Reveals Non-B DNA Structures with Regulatory Potential across a Mammalian Genome. Cell Syst. 2017, 4, 344–356.e7. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Wen, C.; Tang, M.; Duan, R.; Chen, J.; Zhang, J.; Zheng, K.; He, Y.; Hao, Y.; Yu, Q.; et al. G-Quadruplex Structural Variations in Human Genome Associated with Single-Nucleotide Variations and Their Impact on Gene Activity. Proc. Natl. Acad. Sci. USA 2021, 118, e2013230118. [Google Scholar] [CrossRef]
- Lago, S.; Nadai, M.; Cernilogar, F.M.; Kazerani, M.; Domíniguez Moreno, H.; Schotta, G.; Richter, S.N. Promoter G-Quadruplexes and Transcription Factors Cooperate to Shape the Cell Type-Specific Transcriptome. Nat. Commun. 2021, 12, 3885. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, J.; Cuesta, S.M.; Adhikari, S.; Hänsel-Hertsch, R.; Tannahill, D.; Balasubramanian, S. G-Quadruplexes Are Transcription Factor Binding Hubs in Human Chromatin. Genome Biol. 2021, 22, 117. [Google Scholar] [CrossRef]
- Fleming, A.M.; Ding, Y.; Burrows, C.J. Oxidative DNA Damage Is Epigenetic by Regulating Gene Transcription via Base Excision Repair. Proc. Natl. Acad. Sci. USA 2017, 114, 2604–2609. [Google Scholar] [CrossRef] [PubMed]
- Roychoudhury, S.; Pramanik, S.; Harris, H.L.; Tarpley, M.; Sarkar, A.; Spagnol, G.; Sorgen, P.L.; Chowdhury, D.; Band, V.; Klinkebiel, D.; et al. Endogenous Oxidized DNA Bases and APE1 Regulate the Formation of G-Quadruplex Structures in the Genome. Proc. Natl. Acad. Sci. USA 2020, 117, 11409–11420. [Google Scholar] [CrossRef]
- Bhakat, K.K.; Mantha, A.K.; Mitra, S. Transcriptional Regulatory Functions of Mammalian AP-Endonuclease (APE1/Ref-1), an Essential Multifunctional Protein. Antioxid. Redox Signal. 2009, 11, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Seifermann, M.; Epe, B. Oxidatively Generated Base Modifications in DNA: Not Only Carcinogenic Risk Factor but Also Regulatory Mark? Free Radic. Biol. Med. 2017, 107, 258–265. [Google Scholar] [CrossRef]
- Ba, X.; Boldogh, I. 8-Oxoguanine DNA Glycosylase 1: Beyond Repair of the Oxidatively Modified Base Lesions. Redox Biol. 2018, 14, 669–678. [Google Scholar] [CrossRef] [PubMed]
- van Essen, D.; Zhu, Y.; Saccani, S. A Feed-Forward Circuit Controlling Inducible NF-ΚB Target Gene Activation by Promoter Histone Demethylation. Mol. Cell 2010, 39, 750–760. [Google Scholar] [CrossRef]
- Pan, L.; Zhu, B.; Hao, W.; Zeng, X.; Vlahopoulos, S.A.; Hazra, T.K.; Hegde, M.L.; Radak, Z.; Bacsi, A.; Brasier, A.R.; et al. Oxidized Guanine Base Lesions Function in 8-Oxoguanine DNA Glycosylase-1-Mediated Epigenetic Regulation of Nuclear Factor ΚB-Driven Gene Expression. J. Biol. Chem. 2016, 291, 25553–25566. [Google Scholar] [CrossRef]
- Perillo, B.; Ombra, M.N.; Bertoni, A.; Cuozzo, C.; Sacchetti, S.; Sasso, A.; Chiariotti, L.; Malorni, A.; Abbondanza, C.; Avvedimento, E.V. DNA Oxidation as Triggered by H3K9me2 Demethylation Drives Estrogen-Induced Gene Expression. Science 2008, 319, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.W.; Phang, T.; Edwards, M.G.; Geraci, M.W.; Gillespie, M.N. Promoter G-Quadruplex Sequences Are Targets for Base Oxidation and Strand Cleavage during Hypoxia-Induced Transcription. Free Radic. Biol. Med. 2012, 53, 51–59. [Google Scholar] [CrossRef]
- Amente, S.; Bertoni, A.; Morano, A.; Lania, L.; Avvedimento, E.V.; Majello, B. LSD1-Mediated Demethylation of Histone H3 Lysine 4 Triggers Myc-Induced Transcription. Oncogene 2010, 29, 3691–3702. [Google Scholar] [CrossRef]
- Amente, S.; Lania, L.; Avvedimento, E.V.; Majello, B. DNA Oxidation Drives Myc Mediated Transcription. Cell Cycle 2010, 9, 3074–3076. [Google Scholar] [CrossRef] [PubMed]
- Zuchegna, C.; Aceto, F.; Bertoni, A.; Romano, A.; Perillo, B.; Laccetti, P.; Gottesman, M.E.; Avvedimento, E.V.; Porcellini, A. Mechanism of Retinoic Acid-Induced Transcription: Histone Code, DNA Oxidation and Formation of Chromatin Loops. Nucleic Acids Res. 2014, 42, 11040–11055. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, J.; Braganza, A.; Sobol, R.W. Base Excision Repair Facilitates a Functional Relationship Between Guanine Oxidation and Histone Demethylation. Antioxid. Redox Signal. 2013, 18, 2429–2443. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Burrows, C.J. Interplay of Guanine Oxidation and G-Quadruplex Folding in Gene Promoters. J. Am. Chem. Soc. 2020, 142, 1115–1136. [Google Scholar] [CrossRef]
- Tell, G.; Quadrifoglio, F.; Tiribelli, C.; Kelley, M.R. The Many Functions of APE1/Ref-1: Not Only a DNA Repair Enzyme. Antioxid. Redox Signal. 2009, 11, 601–619. [Google Scholar] [CrossRef]
- Fleming, A.M.; Zhu, J.; Ding, Y.; Burrows, C.J. 8-Oxo-7,8-Dihydroguanine in the Context of a Gene Promoter G-Quadruplex Is an On–Off Switch for Transcription. ACS Chem. Biol. 2017, 12, 2417–2426. [Google Scholar] [CrossRef]
- Fleming, A.M.; Zhu, J.; Ding, Y.; Burrows, C.J. Location Dependence of the Transcriptional Response of a Potential G-Quadruplex in Gene Promoters under Oxidative Stress. Nucleic Acids Res. 2019, 47, 5049–5060. [Google Scholar] [CrossRef] [PubMed]
- Yamamori, T.; DeRicco, J.; Naqvi, A.; Hoffman, T.A.; Mattagajasingh, I.; Kasuno, K.; Jung, S.-B.; Kim, C.-S.; Irani, K. SIRT1 Deacetylates APE1 and Regulates Cellular Base Excision Repair. Nucleic Acids Res. 2010, 38, 832–845. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Mantha, A.K.; Song, H.; Roychoudhury, S.; Nath, S.; Ray, S.; Bhakat, K.K. Elevated Level of Acetylation of APE1 in Tumor Cells Modulates DNA Damage Repair. Oncotarget 2016, 7, 75197–75209. [Google Scholar] [CrossRef]
- Cogoi, S.; Ferino, A.; Miglietta, G.; Pedersen, E.B.; Xodo, L.E. The Regulatory G4 Motif of the Kirsten Ras (KRAS) Gene Is Sensitive to Guanine Oxidation: Implications on Transcription. Nucleic Acids Res. 2018, 46, 661–676. [Google Scholar] [CrossRef]
- Zhu, J.; Fleming, A.M.; Burrows, C.J. The RAD17 Promoter Sequence Contains a Potential Tail-Dependent G-Quadruplex That Downregulates Gene Expression upon Oxidative Modification. ACS Chem. Biol 2018, 13, 2577–2584. [Google Scholar] [CrossRef]
- Redstone, S.C.J.; Fleming, A.M.; Burrows, C.J. Oxidative Modification of the Potential G-Quadruplex Sequence in the PCNA Gene Promoter Can Turn on Transcription. Chem. Res. Toxicol. 2019, 32, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Kuninger, D.T.; Izumi, T.; Papaconstantinou, J.; Mitra, S. Human AP-Endonuclease 1 and HnRNP-L Interact with a NCaRE-like Repressor Element in the AP-Endonuclease 1 Promoter. Nucleic Acids Res. 2002, 30, 823–829. [Google Scholar] [CrossRef]
- Antoniali, G.; Lirussi, L.; D’Ambrosio, C.; Dal Piaz, F.; Vascotto, C.; Casarano, E.; Marasco, D.; Scaloni, A.; Fogolari, F.; Tell, G. SIRT1 Gene Expression upon Genotoxic Damage Is Regulated by APE1 through NCaRE-Promoter Elements. Mol. Biol. Cell 2014, 25, 532–547. [Google Scholar] [CrossRef]
- Fleming, A.M.; Zhu, J.; Ding, Y.; Visser, J.A.; Zhu, J.; Burrows, C.J. Human DNA Repair Genes Possess Potential G-Quadruplex Sequences in Their Promoters and 5′-Untranslated Regions. Biochemistry 2018, 57, 991–1002. [Google Scholar] [CrossRef]
- Tian, T.; Chen, Y.-Q.; Wang, S.-R.; Zhou, X. G-Quadruplex: A Regulator of Gene Expression and Its Chemical Targeting. Chem 2018, 4, 1314–1344. [Google Scholar] [CrossRef]
- Robinson, J.; Raguseo, F.; Nuccio, S.P.; Liano, D.; Di Antonio, M. DNA G-Quadruplex Structures: More than Simple Roadblocks to Transcription? Nucleic Acids Res. 2021, 49, 8419–8431. [Google Scholar] [CrossRef]
- Sack, L.M.; Davoli, T.; Li, M.Z.; Li, Y.; Xu, Q.; Naxerova, K.; Wooten, E.C.; Bernardi, R.J.; Martin, T.D.; Chen, T.; et al. Profound Tissue Specificity in Proliferation Control Underlies Cancer Drivers and Aneuploidy Patterns. Cell 2018, 173, 499–514.e23. [Google Scholar] [CrossRef]
- Hänsel-Hertsch, R.; Simeone, A.; Shea, A.; Hui, W.W.I.; Zyner, K.G.; Marsico, G.; Rueda, O.M.; Bruna, A.; Martin, A.; Zhang, X.; et al. Landscape of G-Quadruplex DNA Structural Regions in Breast Cancer. Nat. Genet. 2020, 52, 878–883. [Google Scholar] [CrossRef]
- Lerner, L.K.; Sale, J.E. Replication of G Quadruplex DNA. Genes 2019, 10, 95. [Google Scholar] [CrossRef]
- Bryan, T.M. Mechanisms of DNA Replication and Repair: Insights from the Study of G-Quadruplexes. Molecules 2019, 24, 3439. [Google Scholar] [CrossRef] [PubMed]
- Maffia, A.; Ranise, C.; Sabbioneda, S. From R-Loops to G-Quadruplexes: Emerging New Threats for the Replication Fork. Int. J. Mol. Sci. 2020, 21, 1506. [Google Scholar] [CrossRef] [PubMed]
- Hastings, P.J.; Lupski, J.R.; Rosenberg, S.M.; Ira, G. Mechanisms of Change in Gene Copy Number. Nat. Rev. Genet. 2009, 10, 551–564. [Google Scholar] [CrossRef]
- Paeschke, K.; Bochman, M.L.; Garcia, P.D.; Cejka, P.; Friedman, K.L.; Kowalczykowski, S.C.; Zakian, V.A. Pif1 Family Helicases Suppress Genome Instability at G-Quadruplex Motifs. Nature 2013, 497, 458–462. [Google Scholar] [CrossRef]
- Wu, Y.; Shin-ya, K.; Brosh, R.M., Jr. FANCJ Helicase Defective in Fanconia Anemia and Breast Cancer Unwinds G-Quadruplex DNA To Defend Genomic Stability. Mol. Cell. Biol. 2008, 28, 4116–4128. [Google Scholar] [CrossRef]
- Mendoza, O.; Bourdoncle, A.; Boulé, J.-B.; Brosh, R.M., Jr.; Mergny, J.-L. G-Quadruplexes and Helicases. Nucleic Acids Res. 2016, 44, 1989–2006. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.; Miller, K.M.; Forment, J.V.; Bradshaw, C.R.; Nikan, M.; Britton, S.; Oelschlaegel, T.; Xhemalce, B.; Balasubramanian, S.; Jackson, S.P. Small-Molecule–Induced DNA Damage Identifies Alternative DNA Structures in Human Genes. Nat. Chem. Biol. 2012, 8, 301–310. [Google Scholar] [CrossRef]
- Stork, C.T.; Bocek, M.; Crossley, M.P.; Sollier, J.; Sanz, L.A.; Chédin, F.; Swigut, T.; Cimprich, K.A. Co-Transcriptional R-Loops Are the Main Cause of Estrogen-Induced DNA Damage. eLife 2016, 5, e17548. [Google Scholar] [CrossRef]
- Zhang, X.; Chiang, H.-C.; Wang, Y.; Zhang, C.; Smith, S.; Zhao, X.; Nair, S.J.; Michalek, J.; Jatoi, I.; Lautner, M.; et al. Attenuation of RNA Polymerase II Pausing Mitigates BRCA1-Associated R-Loop Accumulation and Tumorigenesis. Nat. Commun. 2017, 8, 15908. [Google Scholar] [CrossRef]
- Lim, E.; Vaillant, F.; Wu, D.; Forrest, N.C.; Pal, B.; Hart, A.H.; Asselin-Labat, M.-L.; Gyorki, D.E.; Ward, T.; Partanen, A.; et al. Aberrant Luminal Progenitors as the Candidate Target Population for Basal Tumor Development in BRCA1 Mutation Carriers. Nat. Med. 2009, 15, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, G.; Geyer, F.C.; Magnay, F.-A.; McCarthy, A.; Kendrick, H.; Natrajan, R.; MacKay, A.; Grigoriadis, A.; Tutt, A.; Ashworth, A.; et al. BRCA1 Basal-like Breast Cancers Originate from Luminal Epithelial Progenitors and Not from Basal Stem Cells. Cell Stem Cell 2010, 7, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Bach, K.; Pensa, S.; Zarocsinceva, M.; Kania, K.; Stockis, J.; Pinaud, S.; Lazarus, K.A.; Shehata, M.; Simões, B.M.; Greenhalgh, A.R.; et al. Time-Resolved Single-Cell Analysis of Brca1 Associated Mammary Tumourigenesis Reveals Aberrant Differentiation of Luminal Progenitors. Nat. Commun. 2021, 12, 1502. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Suarez, E.; Jacob, A.P.; Jones, J.; Miller, R.; Roudier-Meyer, M.P.; Erwert, R.; Pinkas, J.; Branstetter, D.; Dougall, W.C. RANK Ligand Mediates Progestin-Induced Mammary Epithelial Proliferation and Carcinogenesis. Nature 2010, 468, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Schramek, D.; Leibbrandt, A.; Sigl, V.; Kenner, L.; Pospisilik, J.A.; Lee, H.J.; Hanada, R.; Joshi, P.A.; Aliprantis, A.; Glimcher, L.; et al. Osteoclast Differentiation Factor RANKL Controls Development of Progestin-Driven Mammary Cancer. Nature 2010, 468, 98–102. [Google Scholar] [CrossRef]
- Nolan, E.; Vaillant, F.; Branstetter, D.; Pal, B.; Giner, G.; Whitehead, L.; Lok, S.W.; Mann, G.B.; Rohrbach, K.; Huang, L.-Y.; et al. RANK Ligand as a Potential Target for Breast Cancer Prevention in BRCA1 -Mutation Carriers. Nat. Med. 2016, 22, 933–939. [Google Scholar] [CrossRef]
- Sigl, V.; Owusu-Boaitey, K.; Joshi, P.A.; Kavirayani, A.; Wirnsberger, G.; Novatchkova, M.; Kozieradzki, I.; Schramek, D.; Edokobi, N.; Hersl, J.; et al. RANKL/RANK Control Brca1 Mutation-Driven Mammary Tumors. Cell Res. 2016, 26, 761–774. [Google Scholar] [CrossRef]
- Sau, A.; Lau, R.; Cabrita, M.A.; Nolan, E.; Crooks, P.A.; Visvader, J.E.; Pratt, M.A.C. Persistent Activation of NF-ΚB in BRCA1-Deficient Mammary Progenitors Drives Aberrant Proliferation and Accumulation of DNA Damage. Cell Stem Cell 2016, 19, 52–65. [Google Scholar] [CrossRef]
- Fan, S.; Wang, J.-A.; Yuan, R.; Ma, Y.; Meng, Q.; Erdos, M.R.; Pestell, R.G.; Yuan, F.; Auborn, K.J.; Goldberg, I.D.; et al. BRCA1 Inhibition of Estrogen Receptor Signaling in Transfected Cells. Science 1999, 284, 1354–1356. [Google Scholar] [CrossRef]
- Fan, S.; Ma, Y.X.; Wang, C.; Yuan, R.; Meng, Q.; Wang, J.-A.; Erdos, M.; Goldberg, I.D.; Webb, P.; Kushner, P.J.; et al. Role of Direct Interaction in BRCA1 Inhibition of Estrogen Receptor Activity. Oncogene 2001, 20, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Ma, Y.X.; Wang, C.; Yuan, R.-Q.; Meng, Q.; Wang, J.-A.; Erdos, M.; Goldberg, I.D.; Webb, P.; Kushner, P.J.; et al. P300 Modulates the BRCA1 Inhibition of Estrogen Receptor Activity. Cancer Res. 2002, 62, 141–151. [Google Scholar]
- Pathania, S.; Bade, S.; Le Guillou, M.; Burke, K.; Reed, R.; Bowman-Colin, C.; Su, Y.; Ting, D.T.; Polyak, K.; Richardson, A.L.; et al. BRCA1 Haploinsufficiency for Replication Stress Suppression in Primary Cells. Nat. Commun. 2014, 5, 5496. [Google Scholar] [CrossRef] [PubMed]
- Rennstam, K.; Ringberg, A.; Cunliffe, H.E.; Olsson, H.; Landberg, G.; Hedenfalk, I. Genomic Alterations in Histopathologically Normal Breast Tissue from BRCA1 Mutation Carriers May Be Caused by BRCA1 Haploinsufficiency. Genes Chromosomes Cancer 2010, 49, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Bellacosa, A.; Godwin, A.K.; Peri, S.; Devarajan, K.; Caretti, E.; Vanderveer, L.; Bove, B.; Slater, C.; Zhou, Y.; Daly, M.; et al. Altered Gene Expression in Morphologically Normal Epithelial Cells from Heterozygous Carriers of BRCA1 or BRCA2 Mutations. Cancer Prev. Res. 2010, 3, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Konishi, H.; Mohseni, M.; Tamaki, A.; Garay, J.P.; Croessmann, S.; Karnan, S.; Ota, A.; Wong, H.Y.; Konishi, Y.; Karakas, B.; et al. Mutation of a Single Allele of the Cancer Susceptibility Gene BRCA1 Leads to Genomic Instability in Human Breast Epithelial Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17773–17778. [Google Scholar] [CrossRef]
- Feilotter, H.E.; Michel, C.; Uy, P.; Bathurst, L.; Davey, S. BRCA1 Haploinsufficiency Leads to Altered Expression of Genes Involved in Cellular Proliferation and Development. PLoS ONE 2014, 9, e100068. [Google Scholar] [CrossRef]
- Singh, A.; Xu, Y.-J. The Cell Killing Mechanisms of Hydroxyurea. Genes 2016, 7, 99. [Google Scholar] [CrossRef]
- Papadopoulou, C.; Guilbaud, G.; Schiavone, D.; Sale, J.E. Nucleotide Pool Depletion Induces G-Quadruplex-Dependent Perturbation of Gene Expression. Cell Rep. 2015, 13, 2491–2503. [Google Scholar] [CrossRef]
- Sedic, M.; Skibinski, A.; Brown, N.; Gallardo, M.; Mulligan, P.; Martinez, P.; Keller, P.J.; Glover, E.; Richardson, A.L.; Cowan, J.; et al. Haploinsufficiency for BRCA1 Leads to Cell-Type-Specific Genomic Instability and Premature Senescence. Nat. Commun. 2015, 6, 7505. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. How Does SIRT1 Affect Metabolism, Senescence and Cancer? Nat. Rev. Cancer 2009, 9, 123–128. [Google Scholar] [CrossRef]
- Lin, Z.; Fang, D. The Roles of SIRT1 in Cancer. Genes Cancer 2013, 4, 97–104. [Google Scholar] [CrossRef]
- Wang, R.-H.; Sengupta, K.; Li, C.; Kim, H.-S.; Cao, L.; Xiao, C.; Kim, S.; Xu, X.; Zheng, Y.; Chilton, B.; et al. Impaired DNA Damage Response, Genome Instability, and Tumorigenesis in SIRT1 Mutant Mice. Cancer Cell 2008, 14, 312–323. [Google Scholar] [CrossRef]
- Oberdoerffer, P.; Michan, S.; McVay, M.; Mostoslavsky, R.; Vann, J.; Park, S.-K.; Hartlerode, A.; Stegmuller, J.; Hafner, A.; Loerch, P.; et al. SIRT1 Redistribution on Chromatin Promotes Genomic Stability but Alters Gene Expression during Aging. Cell 2008, 135, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Palacios, J.A.; Herranz, D.; De Bonis, M.L.; Velasco, S.; Serrano, M.; Blasco, M.A. SIRT1 Contributes to Telomere Maintenance and Augments Global Homologous Recombination. J. Cell Biol. 2010, 191, 1299–1313. [Google Scholar] [CrossRef]
- Liu, T.; Liu, P.Y.; Marshall, G.M. The Critical Role of the Class III Histone Deacetylase SIRT1 in Cancer. Cancer Res. 2009, 69, 1702–1705. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-H.; Zheng, Y.; Kim, H.-S.; Xu, X.; Cao, L.; Lahusen, T.; Lee, M.-H.; Xiao, C.; Vassilopoulos, A.; Chen, W.; et al. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-Associated Tumorigenesis. Mol. Cell 2008, 32, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Bi, F.-F.; Chen, N.-N.; Cao, J.-M.; Sun, W.-P.; Zhou, Y.-M.; Li, C.-Y.; Yang, Q. A Novel Crosstalk between BRCA1 and Sirtuin 1 in Ovarian Cancer. Sci. Rep. 2014, 4, 6666. [Google Scholar] [CrossRef]
- Gorrini, C.; Baniasadi, P.S.; Harris, I.S.; Silvester, J.; Inoue, S.; Snow, B.; Joshi, P.A.; Wakeham, A.; Molyneux, S.D.; Martin, B.; et al. BRCA1 Interacts with Nrf2 to Regulate Antioxidant Signaling and Cell Survival. J. Exp. Med. 2013, 210, 1529–1544. [Google Scholar] [CrossRef]
- Savage, K.I.; Matchett, K.B.; Barros, E.M.; Cooper, K.M.; Irwin, G.W.; Gorski, J.J.; Orr, K.S.; Vohhodina, J.; Kavanagh, J.N.; Madden, A.F.; et al. BRCA1 Deficiency Exacerbates Estrogen-Induced DNA Damage and Genomic Instability. Cancer Res. 2014, 74, 2773–2784. [Google Scholar] [CrossRef] [PubMed]
- Yager, J.D. Chapter 3: Endogenous Estrogens as Carcinogens Through Metabolic Activation. J. Natl. Cancer Inst. Monogr. 2000, 2000, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Rajapakse, N.; Butterworth, M.; Kortenkamp, A. Detection of DNA Strand Breaks and Oxidized DNA Bases at the Single-Cell Level Resulting from Exposure to Estradiol and Hydroxylated Metabolites. Environ. Mol. Mutagenesis 2005, 45, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Corominas-Faja, B.; Muñoz-San María, M.; Martin-Castillo, B.; Lupu, R.; Brunet, J.; Bosch-Barrera, J.; Menendez, J.A. BRCA1 Haploinsufficiency Cell-Autonomously Activates RANKL Expression and Generates Denosumab-Responsive Breast Cancer-Initiating Cells. Oncotarget 2017, 8, 35019–35032. [Google Scholar] [CrossRef]
- Saha, T.; Rih, J.K.; Roy, R.; Ballal, R.; Rosen, E.M. Transcriptional Regulation of the Base Excision Repair Pathway by BRCA1. J. Biol. Chem. 2010, 285, 19092–19105. [Google Scholar] [CrossRef]
- Neelsen, K.J.; Lopes, M. Replication Fork Reversal in Eukaryotes: From Dead End to Dynamic Response. Nat. Rev. Mol. Cell Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef]
- Šviković, S.; Sale, J.E. The Effects of Replication Stress on S Phase Histone Management and Epigenetic Memory. J. Mol. Biol. 2017, 429, 2011–2029. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.-Q.; Ghanbarian, A.T.; Spiegel, J.; Martínez Cuesta, S.; Beraldi, D.; Di Antonio, M.; Marsico, G.; Hänsel-Hertsch, R.; Tannahill, D.; Balasubramanian, S. DNA G-Quadruplex Structures Mold the DNA Methylome. Nat. Struct. Mol. Biol. 2018, 25, 951–957. [Google Scholar] [CrossRef]
- Shukla, V.; Coumoul, X.; Lahusen, T.; Wang, R.-H.; Xu, X.; Vassilopoulos, A.; Xiao, C.; Lee, M.-H.; Man, Y.-G.; Ouchi, M.; et al. BRCA1 Affects Global DNA Methylation through Regulation of DNMT1. Cell Res. 2010, 20, 1201–1215. [Google Scholar] [CrossRef]
- Suijkerbuijk, K.P.M.; Fackler, M.J.; Sukumar, S.; van Gils, C.H.; van Laar, T.; van der Wall, E.; Vooijs, M.; van Diest, P.J. Methylation Is Less Abundant in BRCA1-Associated Compared with Sporadic Breast Cancer. Ann. Oncol. 2008, 19, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Vasilatos, S.N.; Broadwater, G.; Barry, W.T.; Baker, J.C.; Lem, S.; Dietze, E.C.; Bean, G.R.; Bryson, A.D.; Pilie, P.G.; Goldenberg, V.; et al. CpG Island Tumor Suppressor Promoter Methylation in Non-BRCA-Associated Early Mammary Carcinogenesis. Cancer Epidemiol. Biomark. Prev. 2009, 18, 901–914. [Google Scholar] [CrossRef]
- Bartlett, T.E.; Chindera, K.; McDermott, J.; Breeze, C.E.; Cooke, W.R.; Jones, A.; Reisel, D.; Karegodar, S.T.; Arora, R.; Beck, S.; et al. Epigenetic Reprogramming of Fallopian Tube Fimbriae in BRCA Mutation Carriers Defines Early Ovarian Cancer Evolution. Nat. Commun. 2016, 7, 11620. [Google Scholar] [CrossRef]
- Tang, M.-H.E.; Varadan, V.; Kamalakaran, S.; Zhang, M.; Dimitrova, N.; Hicks, J. Major Chromosomal Breakpoint Intervals in Breast Cancer Co-Localize with Differentially Methylated Regions. Front. Oncol. 2012, 2, 197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Klinkebiel, D.; Barger, C.J.; Pandey, S.; Guda, C.; Miller, A.; Akers, S.N.; Odunsi, K.; Karpf, A.R. Global DNA Hypomethylation in Epithelial Ovarian Cancer: Passive Demethylation and Association with Genomic Instability. Cancers 2020, 12, 764. [Google Scholar] [CrossRef]
- Mukherjee, A.K.; Sharma, S.; Chowdhury, S. Non-Duplex G-Quadruplex Structures Emerge as Mediators of Epigenetic Modifications. Trends Genet. 2019, 35, 129–144. [Google Scholar] [CrossRef]
- Varizhuk, A.; Isaakova, E.; Pozmogova, G. DNA G-Quadruplexes (G4s) Modulate Epigenetic (Re)Programming and Chromatin Remodeling. BioEssays 2019, 41, 1900091. [Google Scholar] [CrossRef] [PubMed]
- Markowetz, F. A Saltationist Theory of Cancer Evolution. Nat. Genet. 2016, 48, 1102–1103. [Google Scholar] [CrossRef]
- Wang, Y.; Waters, J.; Leung, M.L.; Unruh, A.; Roh, W.; Shi, X.; Chen, K.; Scheet, P.; Vattathil, S.; Liang, H.; et al. Clonal Evolution in Breast Cancer Revealed by Single Nucleus Genome Sequencing. Nature 2014, 512, 155–160. [Google Scholar] [CrossRef]
- Gao, R.; Davis, A.; McDonald, T.O.; Sei, E.; Shi, X.; Wang, Y.; Tsai, P.-C.; Casasent, A.; Waters, J.; Zhang, H.; et al. Punctuated Copy Number Evolution and Clonal Stasis in Triple-Negative Breast Cancer. Nat. Genet. 2016, 48, 1119–1130. [Google Scholar] [CrossRef]
- Zhu, Q.; Pao, G.M.; Huynh, A.M.; Suh, H.; Tonnu, N.; Nederlof, P.M.; Gage, F.H.; Verma, I.M. BRCA1 Tumour Suppression Occurs via Heterochromatin-Mediated Silencing. Nature 2011, 477, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Hoong, N.; Aslanian, A.; Hara, T.; Benner, C.; Heinz, S.; Miga, K.H.; Ke, E.; Verma, S.; Soroczynski, J.; et al. Heterochromatin-Encoded Satellite RNAs Induce Breast Cancer. Mol. Cell 2018, 70, 842–853.e7. [Google Scholar] [CrossRef]
- Gorthi, A.; Romero, J.C.; Loranc, E.; Cao, L.; Lawrence, L.A.; Goodale, E.; Iniguez, A.B.; Bernard, X.; Masamsetti, V.P.; Roston, S.; et al. EWS–FLI1 Increases Transcription to Cause R-Loops and Block BRCA1 Repair in Ewing Sarcoma. Nature 2018, 555, 387–391. [Google Scholar] [CrossRef]
- Stranger, B.E.; Forrest, M.S.; Dunning, M.; Ingle, C.E.; Beazley, C.; Thorne, N.; Redon, R.; Bird, C.P.; de Grassi, A.; Lee, C.; et al. Relative Impact of Nucleotide and Copy Number Variation on Gene Expression Phenotypes. Science 2007, 315, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Henrichsen, C.N.; Vinckenbosch, N.; Zöllner, S.; Chaignat, E.; Pradervand, S.; Schütz, F.; Ruedi, M.; Kaessmann, H.; Reymond, A. Segmental Copy Number Variation Shapes Tissue Transcriptomes. Nat. Genet. 2009, 41, 424–429. [Google Scholar] [CrossRef]
- Fehrmann, R.S.N.; Karjalainen, J.M.; Krajewska, M.; Westra, H.-J.; Maloney, D.; Simeonov, A.; Pers, T.H.; Hirschhorn, J.N.; Jansen, R.C.; Schultes, E.A.; et al. Gene Expression Analysis Identifies Global Gene Dosage Sensitivity in Cancer. Nat. Genet. 2015, 47, 115–125. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Bense, R.D.; Urzúa-Traslaviña, C.G.; de Vries, E.G.E.; van Vugt, M.A.T.M.; Fehrmann, R.S.N. Transcriptional Effects of Copy Number Alterations in a Large Set of Human Cancers. Nat. Commun. 2020, 11, 715. [Google Scholar] [CrossRef]
- Veitia, R.A.; Bottani, S.; Birchler, J.A. Gene Dosage Effects: Nonlinearities, Genetic Interactions, and Dosage Compensation. Trends Genet. 2013, 29, 385–393. [Google Scholar] [CrossRef]
- Beroukhim, R.; Zhang, X.; Meyerson, M. Copy Number Alterations Unmasked as Enhancer Hijackers. Nat. Genet. 2017, 49, 5–6. [Google Scholar] [CrossRef]
- Valton, A.-L.; Dekker, J. TAD Disruption as Oncogenic Driver. Curr. Opin. Genet. Dev. 2016, 36, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Lupiáñez, D.G.; Spielmann, M.; Mundlos, S. Breaking TADs: How Alterations of Chromatin Domains Result in Disease. Trends Genet. 2016, 32, 225–237. [Google Scholar] [CrossRef]
- Spielmann, M.; Lupiáñez, D.G.; Mundlos, S. Structural Variation in the 3D Genome. Nat. Rev. Genet. 2018, 19, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Baluapuri, A.; Wolf, E.; Eilers, M. Target Gene-Independent Functions of MYC Oncoproteins. Nat. Rev. Mol. Cell Biol. 2020, 21, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhang, C.-Z.; Wala, J.; Mermel, C.H.; et al. Pan-Cancer Patterns of Somatic Copy Number Alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Herold, S.; Kalb, J.; Büchel, G.; Ade, C.P.; Baluapuri, A.; Xu, J.; Koster, J.; Solvie, D.; Carstensen, A.; Klotz, C.; et al. Recruitment of BRCA1 Limits MYCN-Driven Accumulation of Stalled RNA Polymerase. Nature 2019, 567, 545–549. [Google Scholar] [CrossRef]
- Tarsounas, M.; Sung, P. The Antitumorigenic Roles of BRCA1–BARD1 in DNA Repair and Replication. Nat. Rev. Mol. Cell Biol. 2020, 21, 284–299. [Google Scholar] [CrossRef]
- Brosh, R.M. DNA Helicases Involved in DNA Repair and Their Roles in Cancer. Nat. Rev. Cancer 2013, 13, 542–558. [Google Scholar] [CrossRef]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 Recruitment to Transcriptional Pause Sites Is Required for R-Loop-Driven DNA Damage Repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef]
- Cohen, S.; Puget, N.; Lin, Y.-L.; Clouaire, T.; Aguirrebengoa, M.; Rocher, V.; Pasero, P.; Canitrot, Y.; Legube, G. Senataxin Resolves RNA:DNA Hybrids Forming at DNA Double-Strand Breaks to Prevent Translocations. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Cristini, A.; Groh, M.; Kristiansen, M.S.; Gromak, N. RNA/DNA Hybrid Interactome Identifies DXH9 as a Molecular Player in Transcriptional Termination and R-Loop-Associated DNA Damage. Cell Rep. 2018, 23, 1891–1905. [Google Scholar] [CrossRef]
- Chakraborty, P.; Hiom, K. DHX9-Dependent Recruitment of BRCA1 to RNA Promotes DNA End Resection in Homologous Recombination. Nat. Commun. 2021, 12, 4126. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Hwang, S. G-Quadruplex Matters in Tissue-Specific Tumorigenesis by BRCA1 Deficiency. Genes 2022, 13, 391. https://doi.org/10.3390/genes13030391
Kim S, Hwang S. G-Quadruplex Matters in Tissue-Specific Tumorigenesis by BRCA1 Deficiency. Genes. 2022; 13(3):391. https://doi.org/10.3390/genes13030391
Chicago/Turabian StyleKim, Sanghyun, and Sohyun Hwang. 2022. "G-Quadruplex Matters in Tissue-Specific Tumorigenesis by BRCA1 Deficiency" Genes 13, no. 3: 391. https://doi.org/10.3390/genes13030391
APA StyleKim, S., & Hwang, S. (2022). G-Quadruplex Matters in Tissue-Specific Tumorigenesis by BRCA1 Deficiency. Genes, 13(3), 391. https://doi.org/10.3390/genes13030391