
Consequences of the Last Glacial Period on the Genetic Diversity of Southeast Asians



Abstract
:1. Introduction
2. Material and Methods
2.1. Genetic Data and Phylogenetic Analysis
2.2. Selection of the Best-Fitting Evolutionary Scenario and Parameters Estimation with Approximate Bayesian Computation
2.2.1. Evolutionary Scenarios and Spatially Explicit Computer Simulations
2.2.2. Summary Statistics
2.2.3. Selection among Alternative Evolutionary Scenarios with Approximate Bayesian Computation
2.2.4. Estimation of Evolutionary Parameters with ABC
3. Results
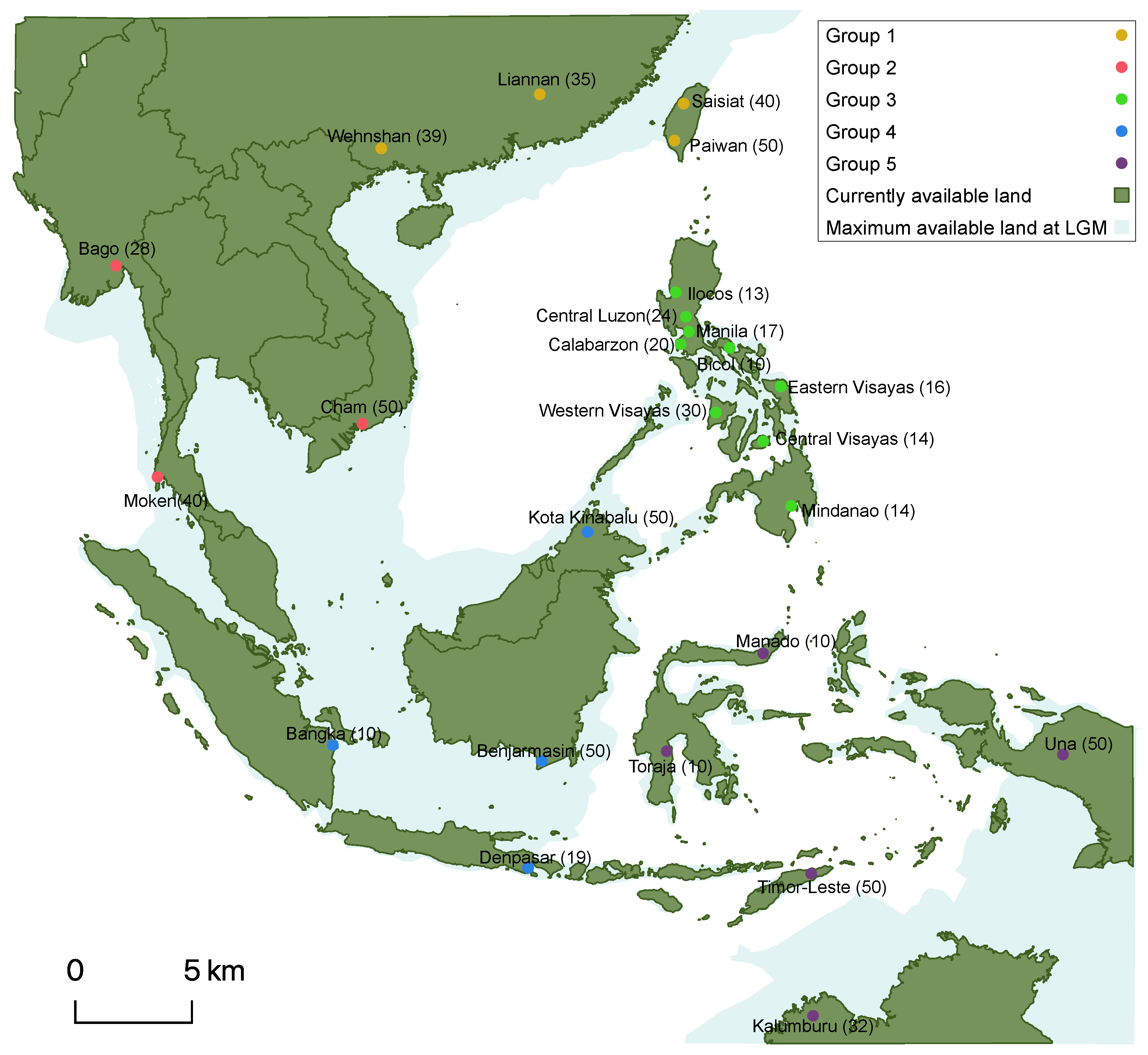
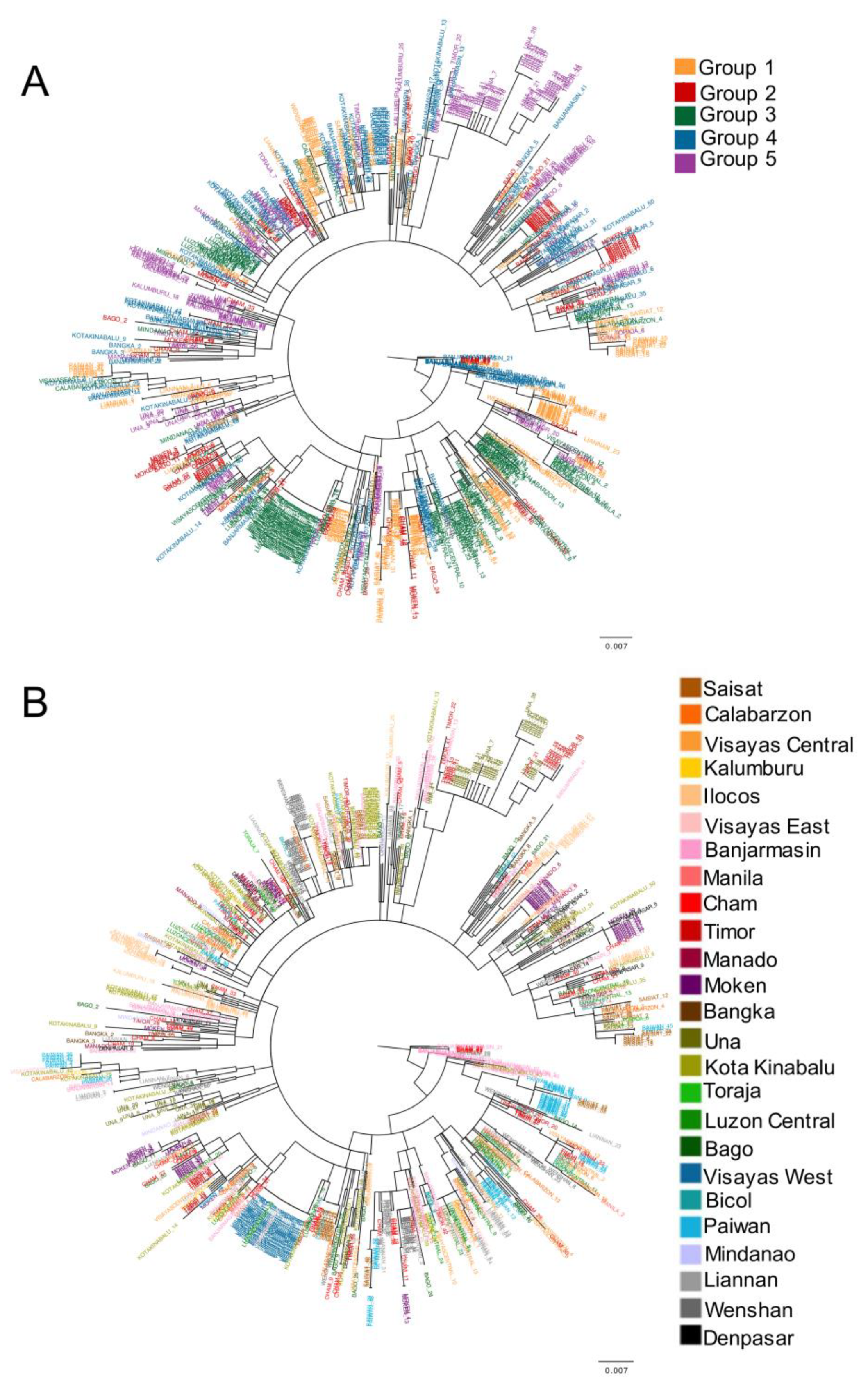
3.1. Phylogenetic Inference Suggests Genetic Admixture between SEA Populations
3.2. The Sea Level Variation Caused by the LGP and LDD Fits with the Observed Genetic Diversity in SEA Populations
3.3. Evolutionary Parameters Estimation Suggests Rapid Migration Favored by Temporarily Exposed Lands Due to the LGP and LDD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bintanja, R.; van de Wal, R.S.W.; Oerlemans, J. Global Ice Volume Variations through the Last Glacial Cycle Simulated by a 3-D Ice-Dynamical Model. Quat. Int. 2002, 95–96, 11–23. [Google Scholar] [CrossRef]
- Stewart, J.R.; Stringer, C.B. Human Evolution Out of Africa: The Role of Refugia and Climate Change. Science 2012, 335, 1317–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arenas, M.; Ray, N.; Currat, M.; Excoffier, L. Consequences of Range Contractions and Range Shifts on Molecular Diversity. Mol. Biol. Evol. 2012, 29, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branco, C.; Ray, N.; Currat, M.; Arenas, M. Influence of Paleolithic Range Contraction, Admixture and Long-Distance Dispersal on Genetic Gradients of Modern Humans in Asia. Mol. Ecol. 2020, 29, 2150–2159. [Google Scholar] [CrossRef]
- Lambeck, K.; Chappell, J. Sea Level Change Through the Last Glacial Cycle. Science 2001, 292, 679–686. [Google Scholar] [CrossRef]
- Melchionna, M.; Di Febbraro, M.; Carotenuto, F.; Rook, L.; Mondanaro, A.; Castiglione, S.; Serio, C.; Vero, V.A.; Tesone, G.; Piccolo, M.; et al. Fragmentation of Neanderthals’ Pre-Extinction Distribution by Climate Change. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2018, 496, 146–154. [Google Scholar] [CrossRef]
- Mondanaro, A.; Di Febbraro, M.; Melchionna, M.; Maiorano, L.; Di Marco, M.; Edwards, N.R.; Holden, P.B.; Castiglione, S.; Rook, L.; Raia, P. The Role of Habitat Fragmentation in Pleistocene Megafauna Extinction in Eurasia. Ecography 2021, 44, 1619–1630. [Google Scholar] [CrossRef]
- Mona, S.; Ray, N.; Arenas, M.; Excoffier, L. Genetic Consequences of Habitat Fragmentation during a Range Expansion. Heredity 2014, 112, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Wren, C.D.; Burke, A. Habitat Suitability and the Genetic Structure of Human Populations during the Last Glacial Maximum (LGM) in Western Europe. PLoS ONE 2019, 14, e0217996. [Google Scholar] [CrossRef] [Green Version]
- Barker, G.; Barton, H.; Bird, M.; Daly, P.; Datan, I.; Dykes, A.; Farr, L.; Gilbertson, D.; Harrisson, B.; Hunt, C.; et al. The ‘Human Revolution’ in Lowland Tropical Southeast Asia: The Antiquity and Behavior of Anatomically Modern Humans at Niah Cave (Sarawak, Borneo). J. Hum. Evol. 2007, 52, 243–261. [Google Scholar] [CrossRef]
- Mijares, A.S.; Détroit, F.; Piper, P.; Grün, R.; Bellwood, P.; Aubert, M.; Champion, G.; Cuevas, N.; De Leon, A.; Dizon, E. New Evidence for a 67,000-Year-Old Human Presence at Callao Cave, Luzon, Philippines. J. Hum. Evol. 2010, 59, 123–132. [Google Scholar] [CrossRef]
- Malaspinas, A.-S.; Westaway, M.C.; Muller, C.; Sousa, V.C.; Lao, O.; Alves, I.; Bergström, A.; Athanasiadis, G.; Cheng, J.Y.; Crawford, J.E.; et al. A Genomic History of Aboriginal Australia. Nature 2016, 538, 207. [Google Scholar] [CrossRef] [PubMed]
- Bird, M.I.; Taylor, D.; Hunt, C. Palaeoenvironments of Insular Southeast Asia during the Last Glacial Period: A Savanna Corridor in Sundaland? Quat. Sci. Rev. 2005, 24, 2228–2242. [Google Scholar] [CrossRef]
- Kealy, S.; Louys, J.; O’Connor, S. Islands Under the Sea: A Review of Early Modern Human Dispersal Routes and Migration Hypotheses Through Wallacea. J. Isl. Coast. Archaeol. 2016, 11, 364–384. [Google Scholar] [CrossRef]
- Voris, H.K. Maps of Pleistocene Sea Levels in Southeast Asia: Shorelines, River Systems and Time Durations. J. Biogeogr. 2000, 27, 1153–1167. [Google Scholar] [CrossRef] [Green Version]
- Alves, I.; Arenas, M.; Currat, M.; Hanulova, A.S.; Sousa, V.C.; Ray, N.; Excoffier, L. Long-Distance Dispersal Shaped Patterns of Human Genetic Diversity in Eurasia. Mol. Biol. Evol. 2016, 33, 946–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arenas, M.; Gorostiza, A.; Baquero, J.M.; Campoy, E.; Branco, C.; Rangel-Villalobos, H.; González-Martín, A. The Early Peopling of the Philippines Based on MtDNA. Sci. Rep. 2020, 10, 4901. [Google Scholar] [CrossRef] [Green Version]
- Brandão, A.; Eng, K.K.; Rito, T.; Cavadas, B.; Bulbeck, D.; Gandini, F.; Pala, M.; Mormina, M.; Hudson, B.; White, J.; et al. Quantifying the Legacy of the Chinese Neolithic on the Maternal Genetic Heritage of Taiwan and Island Southeast Asia. Hum. Genet. 2016, 135, 363–376. [Google Scholar] [CrossRef] [Green Version]
- Hanebuth, T.; Stattegger, K.; Grootes, P.M. Rapid Flooding of the Sunda Shelf: A Late-Glacial Sea-Level Record. Science 2000, 288, 1033–1035. [Google Scholar] [CrossRef]
- Oppenheimer, S. Eden in the East: The Drowned Continent of Southeast Asia; Phoenix, Orion Books Ltd.: London, UK, 1999. [Google Scholar]
- Soares, P.; Trejaut, J.A.; Loo, J.-H.; Hill, C.; Mormina, M.; Lee, C.-L.; Chen, Y.-M.; Hudjashov, G.; Forster, P.; Macaulay, V.; et al. Climate Change and Postglacial Human Dispersals in Southeast Asia. Mol. Biol. Evol. 2008, 25, 1209–1218. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, Q.D.; Gray, R.D.; Drummond, A.J. MtDNA Variation Predicts Population Size in Humans and Reveals a Major Southern Asian Chapter in Human Prehistory. Mol. Biol. Evol. 2008, 25, 468–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macaulay, V.; Hill, C.; Achilli, A.; Rengo, C.; Clarke, D.; Meehan, W.; Blackburn, J.; Semino, O.; Scozzari, R.; Cruciani, F.; et al. Single, Rapid Coastal Settlement of Asia Revealed by Analysis of Complete Mitochondrial Genomes. Science 2005, 308, 1034–1036. [Google Scholar] [CrossRef] [PubMed]
- Tajima, A.; Sun, C.-S.; Pan, I.-H.; Ishida, T.; Saitou, N.; Horai, S. Mitochondrial DNA Polymorphisms in Nine Aboriginal Groups of Taiwan: Implications for the Population History of Aboriginal Taiwanese. Hum. Genet. 2003, 113, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Trejaut, J.A.; Kivisild, T.; Loo, J.H.; Lee, C.L.; He, C.L.; Hsu, C.J.; Li, Z.Y.; Lin, M. Traces of Archaic Mitochondrial Lineages Persist in Austronesian-Speaking Formosan Populations. PLoS Biol. 2005, 3, e247. [Google Scholar]
- Wen, B.; Li, H.; Gao, S.; Mao, X.; Gao, Y.; Li, F.; Zhang, F.; He, Y.; Dong, Y.; Zhang, Y.; et al. Genetic Structure of Hmong-Mien Speaking Populations in East Asia as Revealed by MtDNA Lineages. Mol. Biol. Evol. 2004, 22, 725–734. [Google Scholar] [CrossRef] [Green Version]
- Summerer, M.; Horst, J.; Erhart, G.; Weißensteiner, H.; Schönherr, S.; Pacher, D.; Forer, L.; Horst, D.; Manhart, A.; Horst, B.; et al. Large-Scale Mitochondrial DNA Analysis in Southeast Asia Reveals Evolutionary Effects of Cultural Isolation in the Multi-Ethnic Population of Myanmar. BMC Evol. Biol. 2014, 14, 17. [Google Scholar] [CrossRef] [Green Version]
- Pradutkanchana, S.; Ishida, T.; Kimura, R. Mitochondrial Diversity of the Sea Nomads of Thailand. GenBank Access. 2010, unpublished. [Google Scholar]
- Peng, M.-S.; Quang, H.H.; Dang, K.P.; Trieu, A.V.; Wang, H.-W.; Yao, Y.-G.; Kong, Q.-P.; Zhang, Y.-P. Tracing the Austronesian Footprint in Mainland Southeast Asia: A Perspective from Mitochondrial DNA. Mol. Biol. Evol. 2010, 27, 2417–2430. [Google Scholar] [CrossRef] [Green Version]
- Hill, C.; Soares, P.; Mormina, M.; Macaulay, V.; Clarke, D.; Blumbach, P.B.; Vizuete-Forster, M.; Forster, P.; Bulbeck, D.; Oppenheimer, S.; et al. A Mitochondrial Stratigraphy for Island Southeast Asia. Am. J. Hum. Genet. 2007, 80, 29–43. [Google Scholar] [CrossRef] [Green Version]
- Tommaseo-Ponzetta, M.; Attimonelli, M.; De Robertis, M.; Tanzariello, F.; Saccone, C. Mitochondrial DNA Variability of West New Guinea Populations. Am. J. Phys. Anthropol. 2002, 117, 49–67. [Google Scholar] [CrossRef]
- Gomes, S.M.; Bodner, M.; Souto, L.; Zimmermann, B.; Huber, G.; Strobl, C.; Röck, A.W.; Achilli, A.; Olivieri, A.; Torroni, A.; et al. Human Settlement History between Sunda and Sahul: A Focus on East Timor (Timor-Leste) and the Pleistocenic MtDNA Diversity. BMC Genom. 2015, 16, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudjashov, G.; Kivisild, T.; Underhill, P.A.; Endicott, P.; Sanchez, J.J.; Lin, A.A.; Shen, P.; Oefner, P.; Renfrew, C.; Villems, R.; et al. Revealing the Prehistoric Settlement of Australia by Y Chromosome and MtDNA Analysis. Proc. Natl. Acad. Sci. USA 2007, 104, 8726–8730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinam, T.A.; Hong, L.-C.; Phipps, M.E.; Stoneking, M.; Ameen, M.; Edo, J.; HUGO Pan-Asian SNP Consortium; Saitou, N. Evolutionary History of Continental Southeast Asians: “Early Train” Hypothesis Based on Genetic Analysis of Mitochondrial and Autosomal DNA Data. Mol. Biol. Evol. 2012, 29, 3513–3527. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, A.; Qiao, H.; Zhang, Y.; Shi, W.; Ho, S.Y.; Xu, W.; Zhang, A.; Zhu, C. Performance of Criteria for Selecting Evolutionary Models in Phylogenetics: A Comprehensive Study Based on Simulated Datasets. BMC Evol. Biol. 2010, 10, 242. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Kishino, H.; Yano, T. Dating of the Human-Ape Splitting by a Molecular Clock of Mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A Fast, Scalable and User-Friendly Tool for Maximum Likelihood Phylogenetic Inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [Green Version]
- Currat, M.; Arenas, M.; Quilodran, C.; Excoffier, L.; Ray, N. SPLATCHE3: Simulation of Serial Genetic Data under Spatially Explicit Evolutionary Scenarios Including Long-Distance Dispersal. Bioinformatics 2019, 35, 4480–4483. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Inference of Human Population History from Individual Whole-Genome Sequences. Nature 2011, 475, 493. [Google Scholar] [CrossRef] [Green Version]
- Gravel, S.; Henn, B.M.; Gutenkunst, R.N.; Indap, A.R.; Marth, G.T.; Clark, A.G.; Yu, F.; Gibbs, R.A.; The 1000 Genomes Project; Bustamante, C.D. Demographic History and Rare Allele Sharing among Human Populations. Proc. Natl. Acad. Sci. USA 2011, 108, 11983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marlowe, F.W. Hunter-Gatherers and Human Evolution. Evol. Anthropol. Issues News Rev. 2005, 14, 54–67. [Google Scholar] [CrossRef]
- Sathiamurthy, E.; Voris, H.K. Maps of Holocene Sea Level Transgression and Submerged Lakes on the Sunda Shelf. Trop. Nat. History 2006, 2, 1–44. [Google Scholar]
- Robles, E.; Piper, P.; Ochoa, J.; Lewis, H.; Paz, V.; Ronquillo, W. Late Quaternary Sea-Level Changes and the Palaeohistory of Palawan Island, Philippines. J. Isl. Coast. Archaeol. 2015, 10, 76–96. [Google Scholar] [CrossRef] [Green Version]
- Ray, N.; Excoffier, L. A First Step towards Inferring Levels of Long-Distance Dispersal during Past Expansions. Mol. Ecol. Resour. 2010, 10, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Soares, P.; Ermini, L.; Thomson, N.; Mormina, M.; Rito, T.; Röhl, A.; Salas, A.; Oppenheimer, S.; Macaulay, V.; Richards, M.B. Correcting for Purifying Selection: An Improved Human Mitochondrial Molecular Clock. Am. J. Hum. Genet. 2009, 84, 740–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Excoffier, L.; Lischer, H.E.L. Arlequin Suite Ver 3.5: A New Series of Programs to Perform Population Genetics Analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Csilléry, K.; François, O.; Blum, M.G.B. Abc: An R Package for Approximate Bayesian Computation (ABC). Methods Ecol. Evol. 2012, 3, 475–479. [Google Scholar] [CrossRef] [Green Version]
- Wegmann, D.; Leuenberger, C.; Neuenschwander, S.; Excoffier, L. ABCtoolbox: A Versatile Toolkit for Approximate Bayesian Computations. BMC Bioinform. 2010, 11, 116. [Google Scholar] [CrossRef] [Green Version]
- Pimenta, J.; Lopes, A.M.; Comas, D.; Amorim, A.; Arenas, M. Evaluating the Neolithic Expansion at Both Shores of the Mediterranean Sea. Mol. Biol. Evol. 2017, 34, 3232–3242. [Google Scholar] [CrossRef]
- Madrigal, L.; Posthumously, L.C.; Melendez-Obando, M.; Villegas-Palma, R.; Barrantes, R.; Raventos, H.; Pereira, R.; Luiselli, D.; Pettener, D.; Barbujani, G. High Mitochondrial Mutation Rates Estimated from Deep-Rooting Costa Rican Pedigrees. Am. J. Phys. Anthropol. 2012, 148, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, S. Paleocene–Eocene and Plio–Pleistocene Sea-Level Changes as “Species Pumps” in Southeast Asia: Evidence from Althepus Spiders. Mol. Phylogenet. Evol. 2018, 127, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Hisheh, S.; Westerman, M.; Schmitt, L.H. Biogeography of the Indonesian Archipelago: Mitochondrial DNA Variation in the Fruit Bat, Eonycteris Spelaea. Biol. J. Linn. Soc. 1998, 65, 329–345. [Google Scholar]
- Consequences of the Last Glacial Period on the Genetic Diversity of Southeast Asians. Available online: http://doi.org/10.5281/zenodo.5515856 (accessed on 12 February 2022).
- Epperson, B.K.; Mcrae, B.H.; Scribner, K.; Cushman, S.A.; Rosenberg, M.S.; Fortin, M.-J.; James, P.M.A.; Murphy, M.; Manel, S.; Legendre, P.; et al. Utility of Computer Simulations in Landscape Genetics. Mol. Ecol. 2010, 19, 3549–3564. [Google Scholar] [CrossRef] [PubMed]
- Benguigui, M.; Arenas, M. Spatial and Temporal Simulation of Human Evolution. Methods, Frameworks and Applications. Curr. Genom. 2014, 15, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Leempoel, K.; Duruz, S.; Rochat, E.; Widmer, I.; Orozco-terWengel, P.; Joost, S. Simple Rules for an Efficient Use of Geographic Information Systems in Molecular Ecology. Front. Ecol. Evol. 2017, 5, 33. [Google Scholar] [CrossRef] [Green Version]
- Kimura, M.; Weiss, G.H. The Stepping Stone Model of Population Structure and the Decrease of Genetic Correlation with Distance. Genetics 1964, 49, 561–576. [Google Scholar] [CrossRef]
- Hill, C.; Soares, P.; Mormina, M.; Macaulay, V.; Meehan, W.; Blackburn, J.; Clarke, D.; Raja, J.M.; Ismail, P.; Bulbeck, D.; et al. Phylogeography and Ethnogenesis of Aboriginal Southeast Asians. Mol. Biol. Evol. 2006, 23, 2480–2491. [Google Scholar] [CrossRef] [Green Version]
- Oppenheimer, S. Out-of-Africa, the Peopling of Continents and Islands: Tracing Uniparental Gene Trees across the Map. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 770–784. [Google Scholar] [CrossRef] [Green Version]
- Arenas, M.; François, O.; Currat, M.; Ray, N.; Excoffier, L. Influence of Admixture and Paleolithic Range Contractions on Current European Diversity Gradients. Mol. Biol. Evol. 2013, 30, 57–61. [Google Scholar] [CrossRef] [Green Version]
- Henn, B.M.; Gignoux, C.R.; Feldman, M.W.; Mountain, J.L. Characterizing the Time Dependency of Human Mitochondrial DNA Mutation Rate Estimates. Mol. Biol. Evol. 2009, 26, 217–230. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Evaluated Evolutionary Scenarios | Posterior Probability | |||||||
|---|---|---|---|---|---|---|---|---|
| Mnlogistic | Neuralnet | |||||||
| (A) NONE vs. LGP vs. LDD vs. LGP&LDD | 0.000 | 0.000 | 0.130 | 0.869 | 0.002 | 0.003 | 0.130 | 0.701 |
| (B) LDD vs. LGP | 0.989 | 0.011 | 1.000 | 0.000 | ||||
| (C) LGP vs. LGP&LDD | 0.001 | 0.999 | 0.001 | 0.999 | ||||
| (D) LDD vs. LGP&LDD | 0.091 | 0.909 | 0.171 | 0.829 | ||||
| Parameter | Mode | Mean | Median | 95% HPDI |
|---|---|---|---|---|
| Time of onset of the expansion(T) | 64,650 † | 64,900 † | 64,875 † | 60,325–69,475 † |
| Population size at the onset of the expansion (N) | 37,462 | 48,570 | 48,173 | 25,025–70,606 |
| Population growth rate (r) | 0.5223 | 0.671 | 0.659 | 0.400–0.946 |
| Migration rate (m) | 0.222 | 0.246 | 0.243 | 0.200–0.291 |
| Migration rate in temporary lands (m_temp) | 0.219 | 0.247 | 0.246 | 0.200–0.292 |
| Carrying capacity (K) | 1849 | 2387 | 2336 | 1001–3727 |
| Carrying capacity in temporary lands (K_temp) | 2660 | 2466 | 2465 | 1049–3794 |
| Mutation rate (μ) | 3.904 × 10−6 | 4.759 × 10−6 | 4.665 × 10−6 | 1.051 × 10−7–9.213 × 10−6 |
| LDD proportion (LDDprop) | 0.027 | 0.029 | 0.029 | 0.011–0.047 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Branco, C.; Kanellou, M.; González-Martín, A.; Arenas, M. Consequences of the Last Glacial Period on the Genetic Diversity of Southeast Asians. Genes 2022, 13, 384. https://doi.org/10.3390/genes13020384
Branco C, Kanellou M, González-Martín A, Arenas M. Consequences of the Last Glacial Period on the Genetic Diversity of Southeast Asians. Genes. 2022; 13(2):384. https://doi.org/10.3390/genes13020384
Chicago/Turabian StyleBranco, Catarina, Marina Kanellou, Antonio González-Martín, and Miguel Arenas. 2022. "Consequences of the Last Glacial Period on the Genetic Diversity of Southeast Asians" Genes 13, no. 2: 384. https://doi.org/10.3390/genes13020384
APA StyleBranco, C., Kanellou, M., González-Martín, A., & Arenas, M. (2022). Consequences of the Last Glacial Period on the Genetic Diversity of Southeast Asians. Genes, 13(2), 384. https://doi.org/10.3390/genes13020384