
The Complete Chloroplast Genome of Carya cathayensis and Phylogenetic Analysis

Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Extraction, Sequencing, and cp Genome Assembly
2.2. Annotation of the C. cathayensis cp Genome
2.3. Identification of Repeats
2.4. Phylogenetic Analysis
2.5. Genomic Comparison with Related Species
3. Results
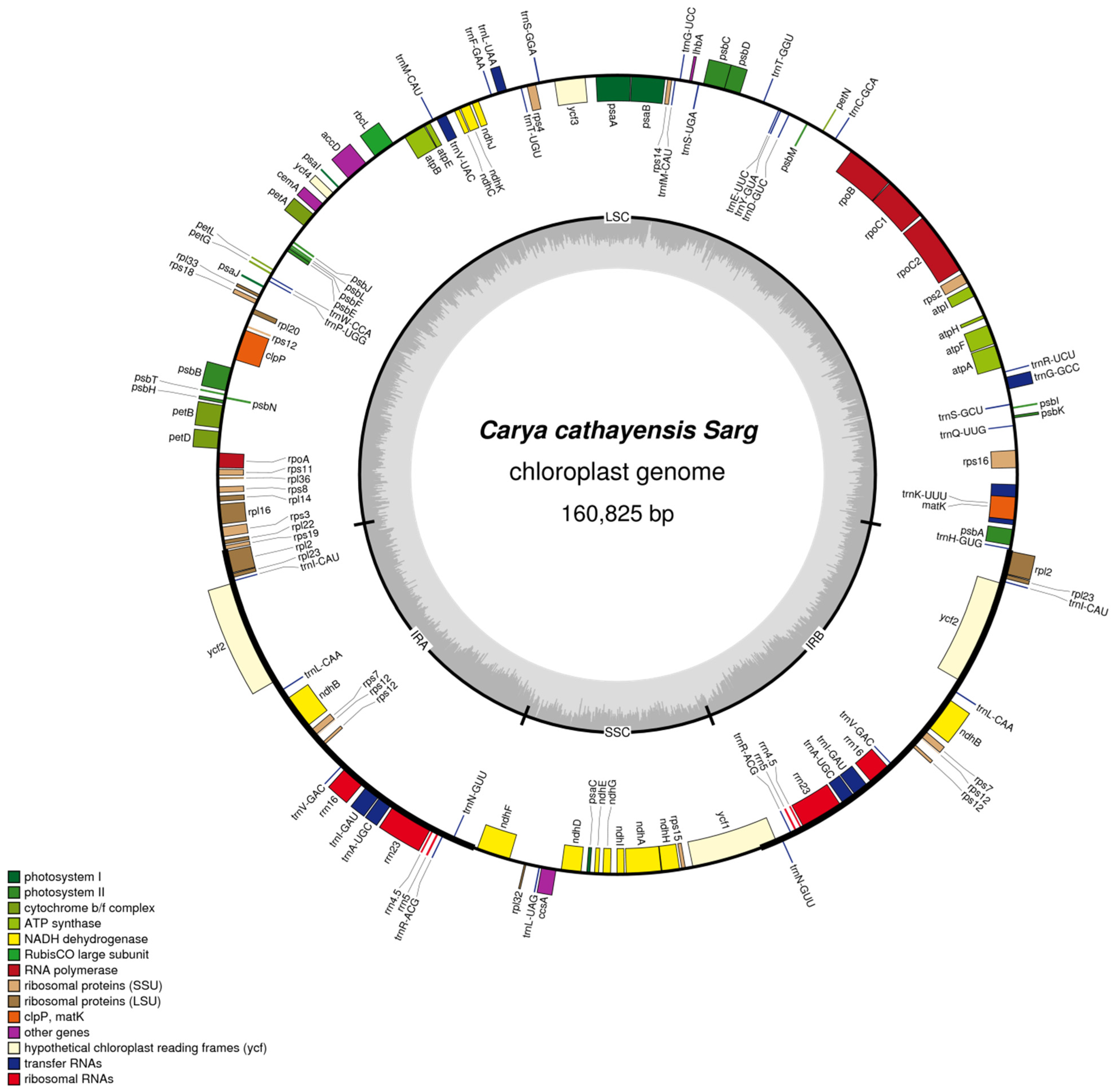
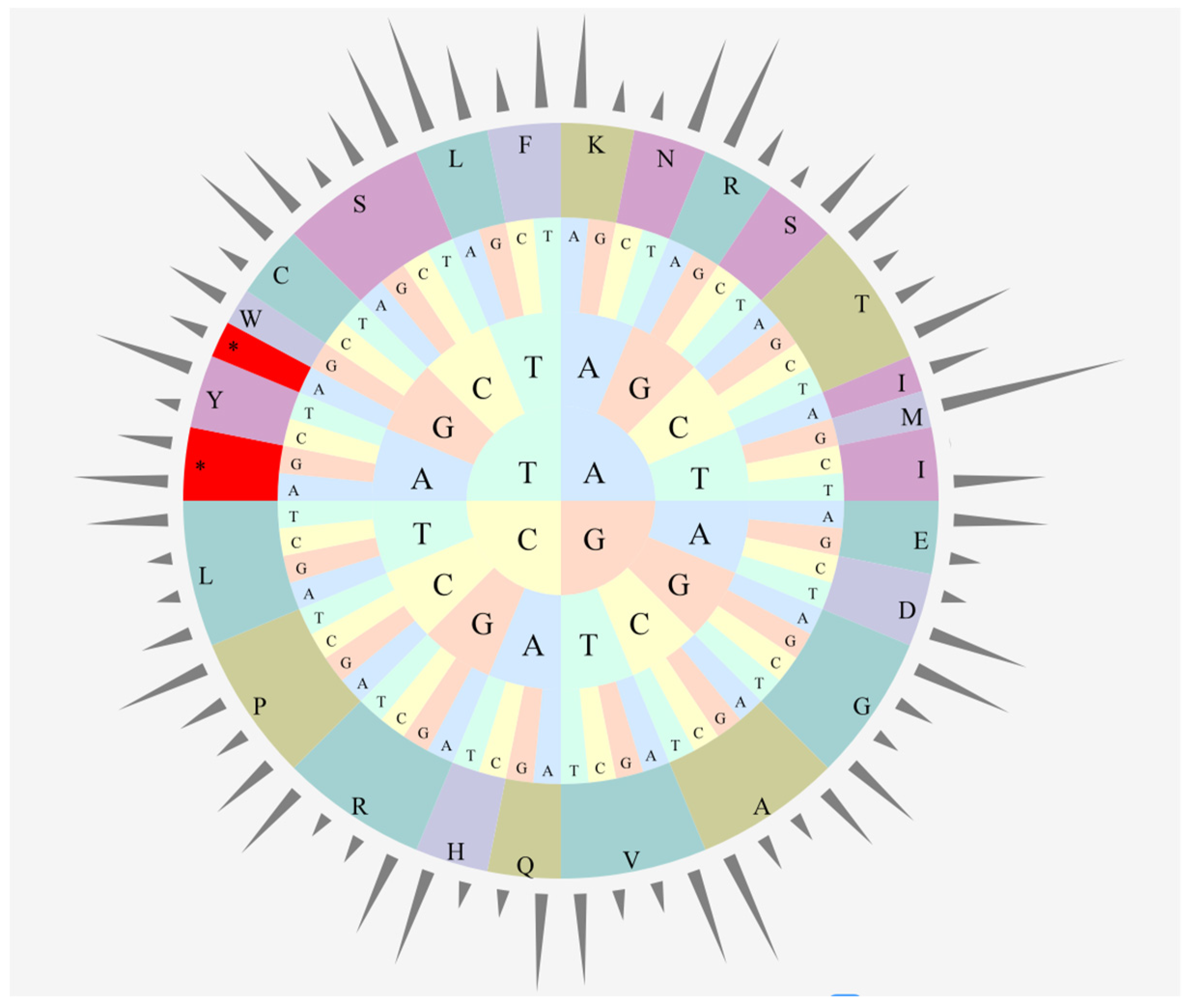
3.1. Genome Features of C. cathayensis
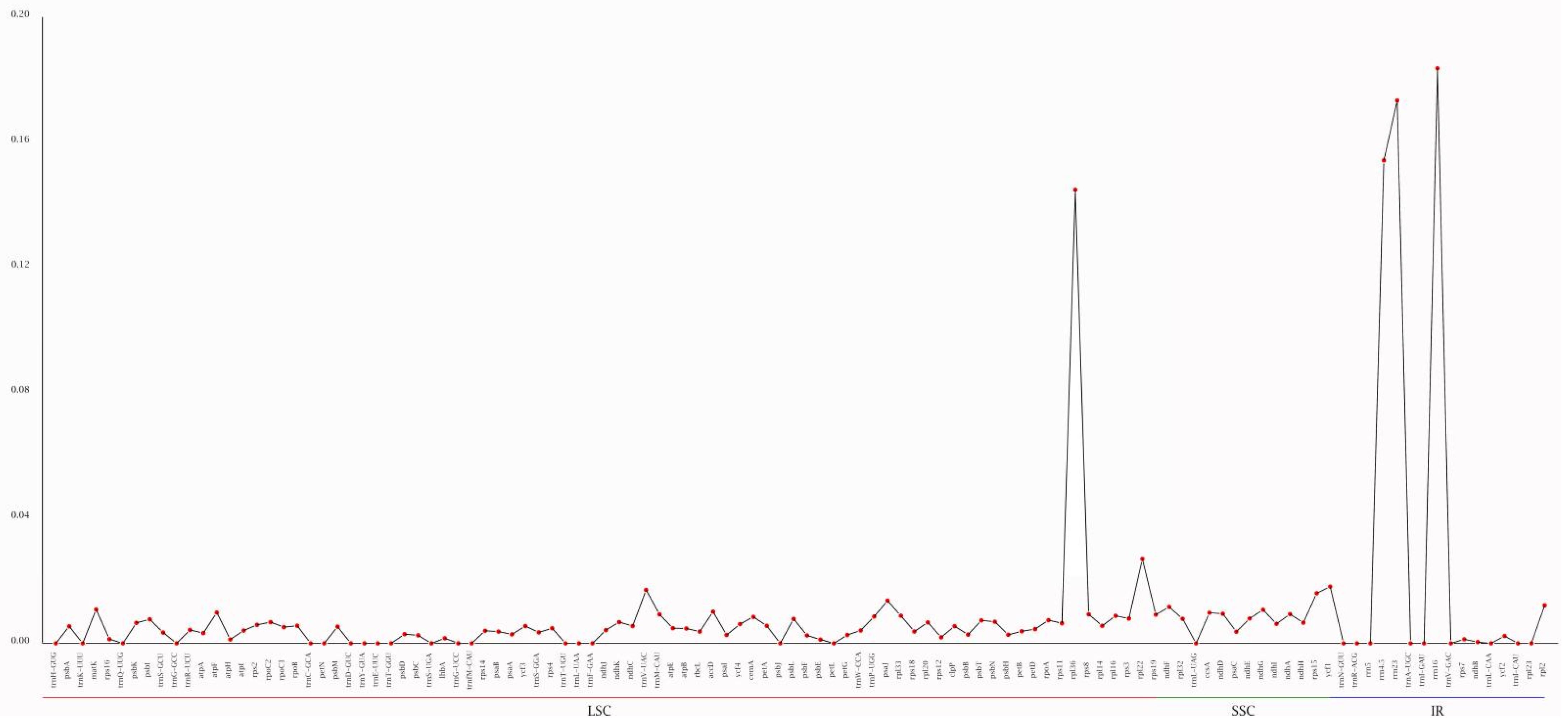
3.2. Analysis of Long Repeats and Simple Sequence Repeats (SSRs)
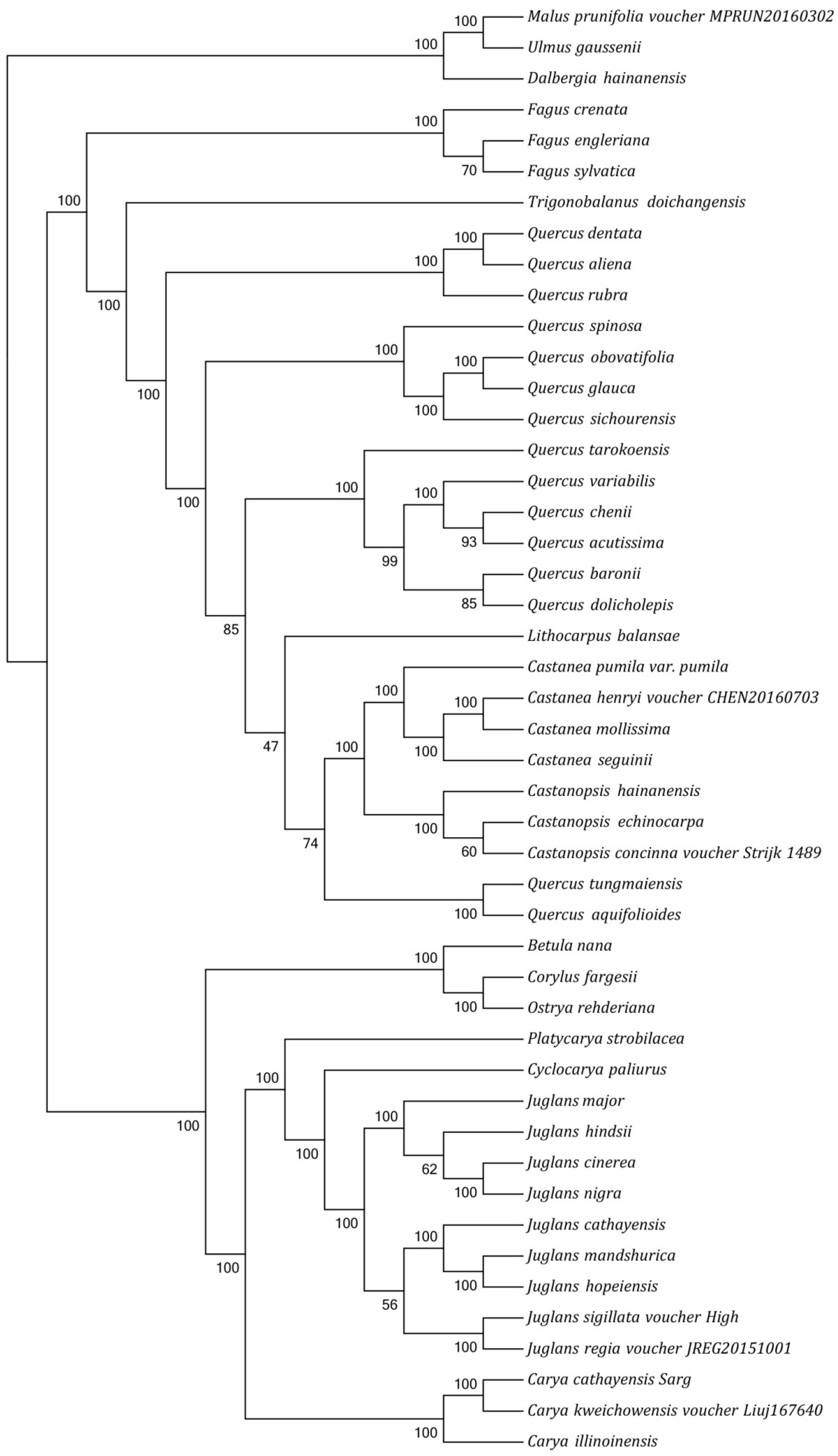
3.3. Phylogenetic Analysis
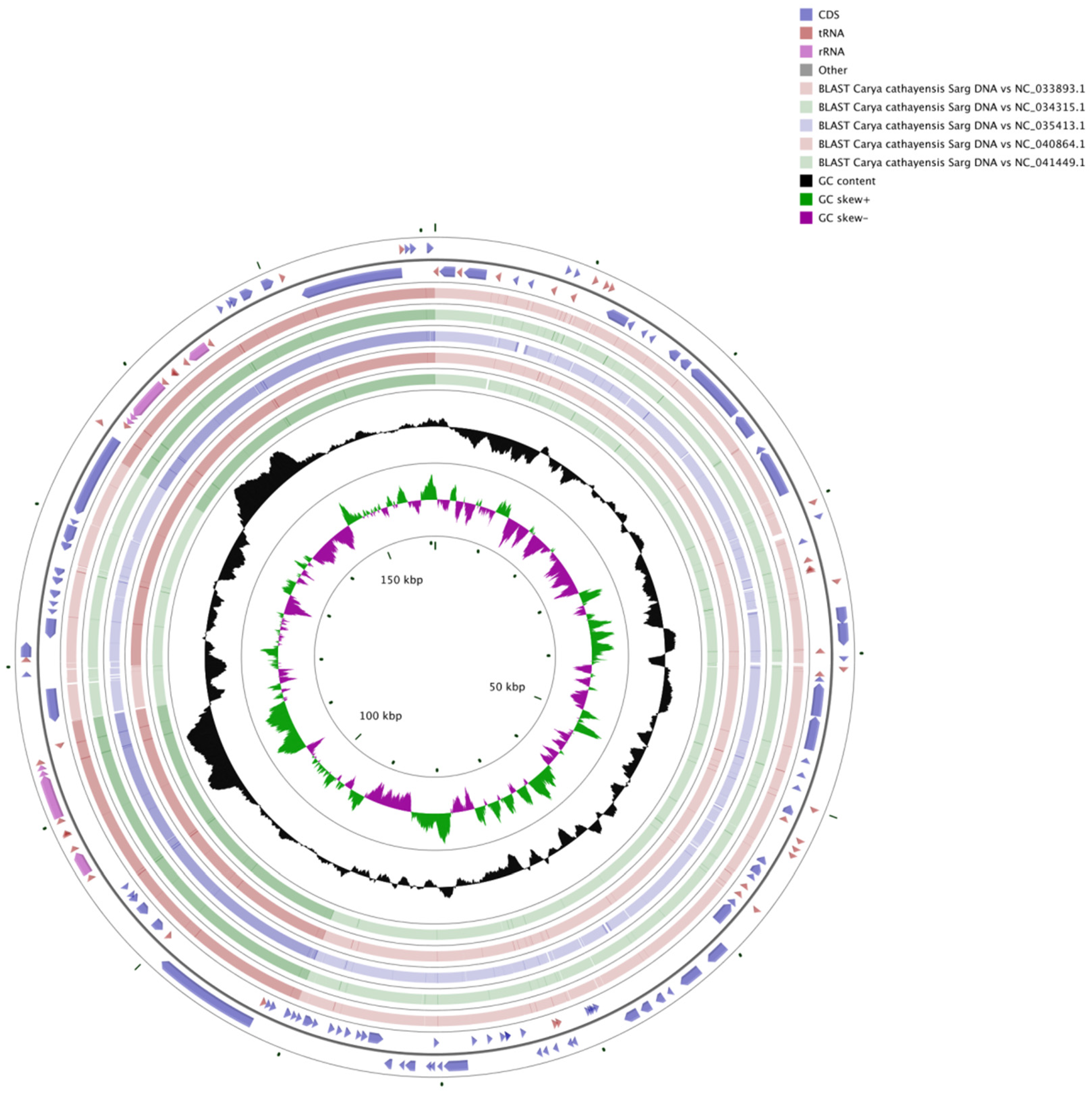
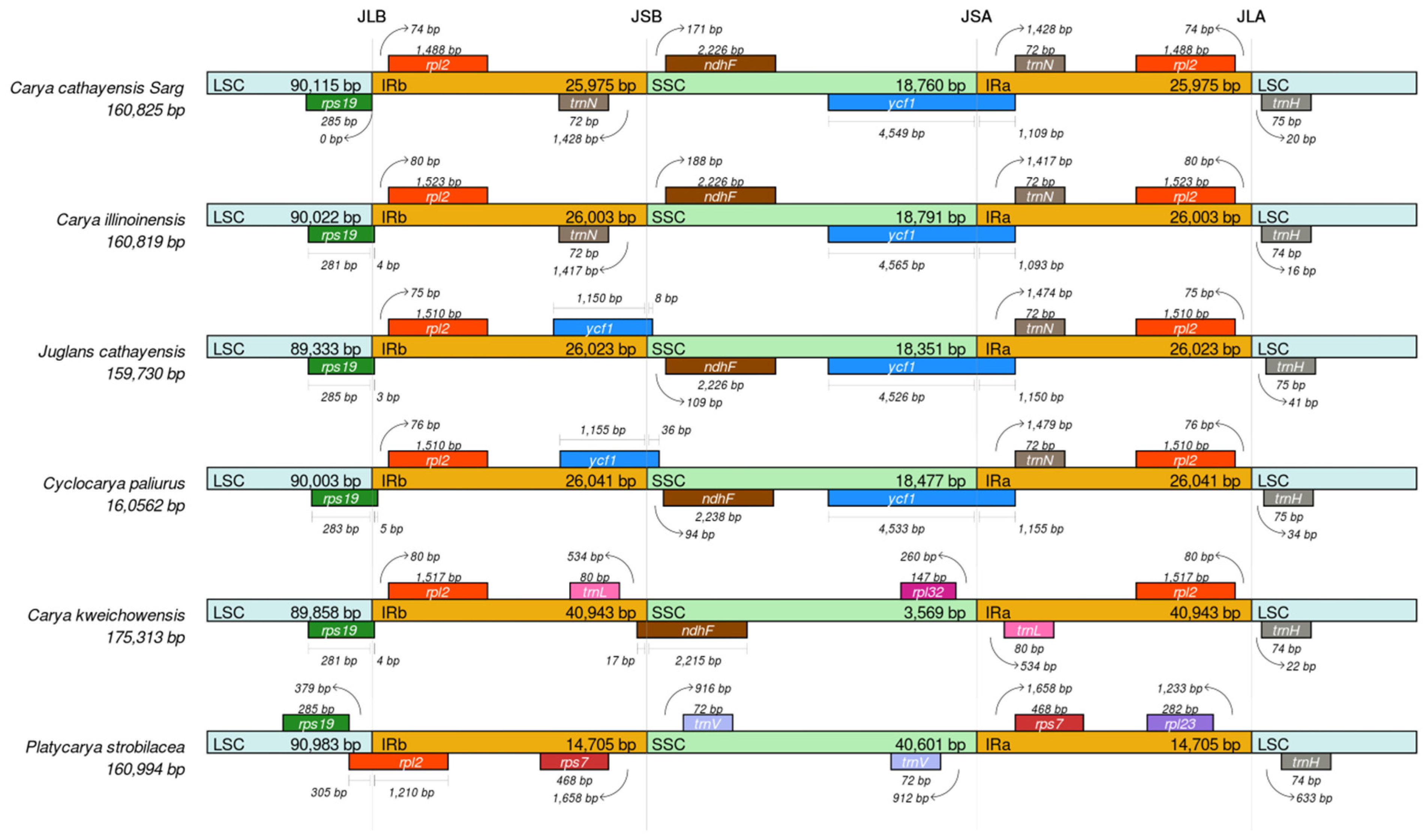
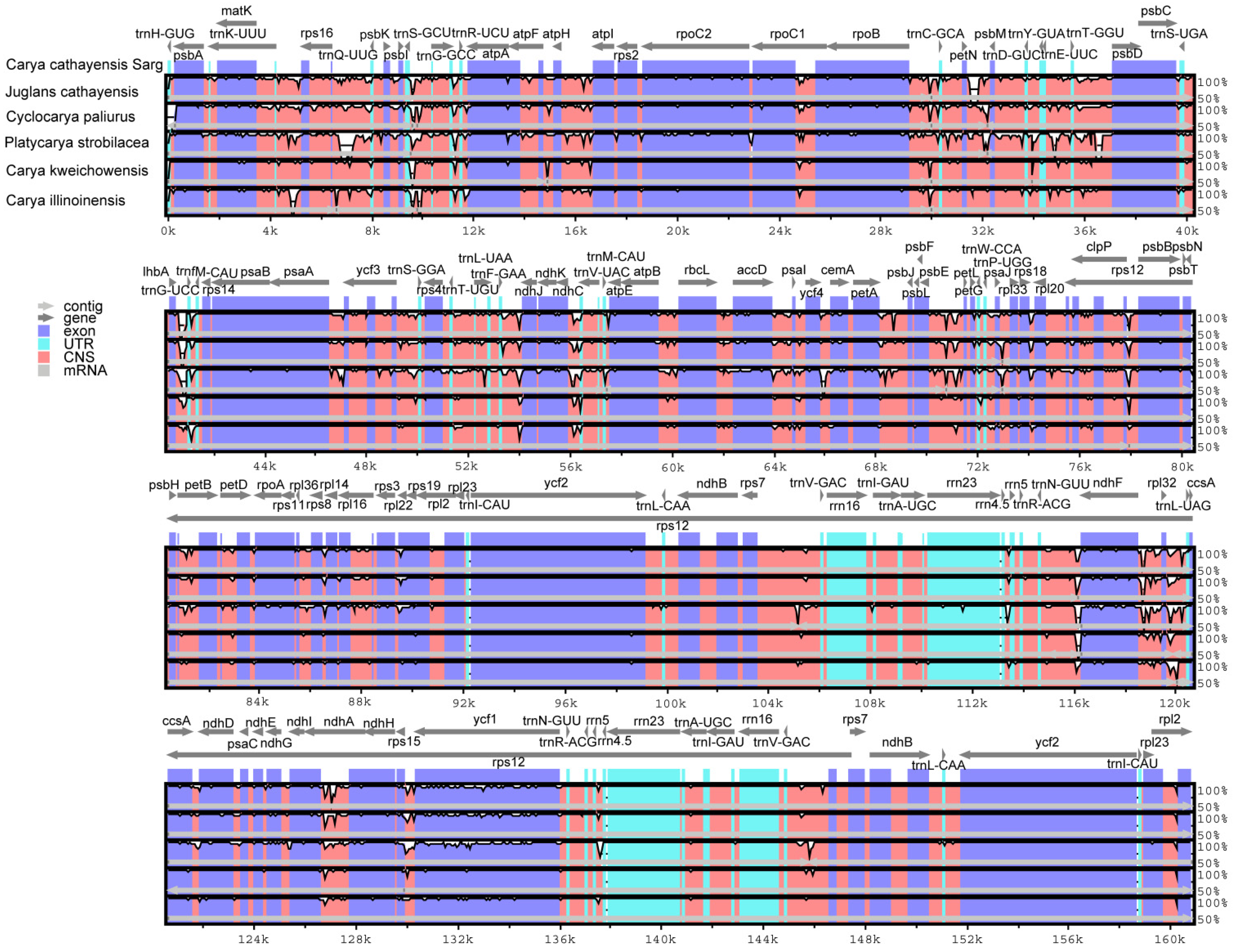
3.4. Comparative Analysis of Genome Structure
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lu, A.; Stone, D.; Grauke, L. Juglandaceae. Flora China 1999, 4, 277–285. [Google Scholar]
- Zhang, J.-B.; Li, R.-Q.; Xiang, X.-G.; Manchester, S.R.; Lin, L.; Wang, W.; Wen, J.; Chen, Z.-D. Integrated fossil and molecular data reveal the biogeographic diversification of the Eastern Asian-Eastern North American Disjunct Hickory genus (Carya Nutt.). PLoS ONE 2013, 8, e70449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naumann, J.; Symmank, L.; Samain, M.S.; Kai, F.M.; Wanke, S. Chasing the hare—Evaluating the phylogenetic utility of a nuclear single copy gene region at and below species level within the species rich group Peperomia (Piperaceae). BMC Evol. Biol. 2011, 11, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raman, G.; Park, V.; Kwak, M.; Lee, B.; Park, S.J. Characterization of the complete chloroplast genome of Arabis stellari and comparisons with related species. PLoS ONE 2017, 12, e0183197. [Google Scholar] [CrossRef] [PubMed]
- Böhle, U.R.; Hilger, H.; Cerff, R.; Martin, W. Noncoding chloroplast DNA for plant molecular systematics at the infrageneric level. In Molecular Ecology and Evolution: Approaches and Applications; Schierwater, B., Streit, G.P., Desalle, R., Eds.; Birkhäuser: Basel, Switzerland, 1994; pp. 391–403. [Google Scholar]
- Li, X.; Li, Y.; Zang, M.; Li, M.; Fang, Y. Complete chloroplast genome sequence and phylogenetic analysis of Quercus acutissima. Int. J. Mol. Sci. 2018, 19, 2443. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Sylvester, S.P.; Li, M.; Zhang, C.; Li, X.; Duan, Y.; Wang, X. The complete plastid genome of Magnolia zenii and genetic comparison to Magnoliaceae species. Molecules 2019, 24, 261. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Xu, Y.; Xi, L.; Yang, J.; Chen, H.; Zhang, J. Characterization of the chloroplast genome sequence of Acer miaotaiense: Comparative and phylogenetic analyses. Molecules 2018, 23, 1740. [Google Scholar] [CrossRef] [Green Version]
- Zeng, S.; Zhou, T.; Han, K.; Yang, Y.; Zhao, J.; Liu, Z.L. The complete chloroplast genome sequences of six Rehmannia species. Genes 2017, 8, 103. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Dong, W.; Li, W.; Lu, Y.; Xie, X.; Jin, X.; Shi, J.; He, K.; Suo, Z. Comparative analysis of six Lagerstroemia complete chloroplast genomes. Front. Plant Sci. 2017, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Fu, C.; Wang, Y.; Liu, J.; Gao, L. Characterization of the complete plastid genome of a Chinese endemic species Carya kweichowensis. Mitochondrial DNA Part B 2018, 3, 492–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, D.-C.; Yao, Q.; Cao, X.-F.; Hao, Q.-Q.; Ma, M.-T.; Pan, J.; Bai, X.-H. Complete chloroplast genome of the wild-type Hickory (Carya cathayensis). Mitochondrial DNA Part B 2019, 4, 1457–1458. [Google Scholar] [CrossRef]
- Zhang, R.; Peng, F.; Li, Y. Pecan production in China. Sci. Hortic. 2015, 197, 719–727. [Google Scholar] [CrossRef]
- Grauke, L.J.; Wood, B.W.; Harris, M.K. Crop vulnerability: Carya. Hortscience 2016, 51, 653–663. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.H.; Huang, J.Q.; Li, X.Q.; Zheng, B.S.; Wu, J.S.; Wang, Z.J.; Liu, G.H.; Chen, M. Effects of potassium supply on limitations of photosynthesis by mesophyll diffusion conductance in Carya cathayensis. Tree Physiol. 2011, 31, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Xiao, L.; Zhang, Z.; Zhang, R.; Wang, Z.; Huang, C.; Huang, R.; Luan, Y.; Fan, T.; Wang, J.; et al. The genomes of pecan and Chinese hickory provide insights into Carya evolution and nut nutrition. Gigascience 2019, 8, giz036. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Shi, L.; Zhu, Y.; Chen, H.; Zhang, J.; Lin, X.; Guan, X. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genom. 2012, 13, 715. [Google Scholar] [CrossRef] [Green Version]
- Kurt, S. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2011, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, S.; Schleiermacher, C. REPuter: Fast computation of maximal repeats in complete genomes. Bioinformatics 1999, 15, 426–427. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.Y.; Yu, Y.; Deng, Y.Q.; Li, J.; Huang, Z.X.; Zhou, S.D. The chloroplast genome of Lilium henrici: Genome structure and comparative analysis. Molecules 2018, 23, 1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Wang, Y.; He, P.; Li, P.; Lee, J.; Soltis, D.E.; Fu, C. Chloroplast genome analyses and genomic resource development for epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae), using genome skimming data. BMC Genomics 2018, 19, 235. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, L.H.; Huang, J.X.; Zhang, G.Q.; Liu, Z.J.; Zhuang, X.Y. A molecular phylogeny of Aeridinae (Orchidaceae: Epidendroideae) inferred from multiple nuclear and chloroplast regions. Mol. Phylogenet. Evol. 2015, 85, 247–254. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Kuraku, S.; Zmasek, C.M.; Nishimura, O.; Katoh, K. aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 2013, 41, W22–W28. [Google Scholar] [CrossRef] [Green Version]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.; Pachter, L.S.; Dubchak, I. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 2000, 16, 1046–1047. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; Depamphilis, C.W.; Chumley, T.W.; Haberle, R.C.; Wyman, S.K.; Alverson, A.J.; Peery, R.; Herman, S.J.; et al. Methods for obtaining and analyzing whole chloroplast genome sequences. Method Enzymol. 2005, 395, 348. [Google Scholar]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Chen, X.; Feng, X.; Woeste, K.E.; Zhao, P. Characterization of the complete chloroplast genome of the endangered species Carya sinensis (Juglandaceae). Conserv. Genet. Resour. 2016, 8, 467–470. [Google Scholar] [CrossRef]
- Morton, B.R. The role of context-dependent mutations in generating compositional and codon usage bias in grass chloroplast DNA. J. Mol. Evol. 2003, 56, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Necsulea, A.; Lobry, J. A new method for assessing the effect of replication on DNA base composition asymmetry. Mol. Biol. Evol. 2007, 24, 2169–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, H.-Y.; Zhang, Y.-H.; Yan, H.-J.; Qiu, X.-Q.; Wang, Q.-G.; Li, S.-B.; Zhang, S.-D. The complete chloroplast genome of a key ancestor of modern Roses, Rosa chinensis var. spontanea, and a comparison with congeneric species. Molecules 2018, 23, 389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.; Wu, M.; Liao, B.; Liu, Z.; Bai, R.; Xiao, S.; Li, X.; Zhang, B.; Xu, J.; Chen, S. Complete chloroplast genome sequence and phylogenetic analysis of the medicinal plant Artemisia annua. Molecules 2017, 22, 1330. [Google Scholar] [CrossRef]
- Ebert, D.; Peakall, R. Chloroplast simple sequence repeats (cpSSRs): Technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species. Mol. Ecol. Resour. 2009, 9, 673–690. [Google Scholar] [CrossRef]
- Provan, J.; Powell, W.; Hollingsworth, P.M. Chloroplast microsatellites: New tools for studies in plant ecology and evolution. Trends Ecol. Evol. 2011, 16, 142–147. [Google Scholar] [CrossRef]
- Diekmann, K.; Hodkinson, T.R.; Barth, S. New chloroplast microsatellite markers suitable for assessing genetic diversity of Lolium perenne and other related grass species. Ann. Bot. 2012, 110, 1327–1339. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Pal, A.K.; Roy, R.K.; Tamta, S.; Rana, T.S. Development of cpSSR markers for analysis of genetic diversity in Gladiolus cultivars. Plant Gene 2017, 10, 31–36. [Google Scholar] [CrossRef]
- Hu, J.B.; Li, J.W.; Zhou, X.Y. Analysis of cytoplasmic variation in a cucumber germplasm collection using chloroplast microsatellite markers. Acta Physiol. Plant 2009, 31, 1085–1089. [Google Scholar] [CrossRef]
- Deng, Q.; Zhang, H.; He, Y.; Wang, T.; Sun, Y. Chloroplast microsatellite markers for Pseudotaxus chienii developed from the whole chloroplast genome of Taxus chinensis var. Mairei (Taxaceae). Appl. Plant Sci. 2017, 5, 1600153. [Google Scholar] [CrossRef]
- Pan, L.; Li, Y.; Guo, R.; Wu, H.; Hu, Z.; Chen, C. Development of 12 chloroplast microsatellite markers in Vigna unguiculata (Fabaceae) and amplification in Phaseolus vulgaris. Appl. Plant Sci. 2014, 2, 1300075. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yang, X.; Zhang, C.; Yin, X.; Liu, S.; Li, X. Development of chloroplast microsatellite markers and analysis of chloroplast diversity in Chinese Jujube (Ziziphus jujuba Mill.) and Wild Jujube (Ziziphus acidojujuba Mill.). PLoS ONE 2015, 10, e0134519. [Google Scholar] [CrossRef]
- Mo, Z.; Lou, W.; Chen, Y.; Jia, X.; Zhai, M.; Guo, Z.; Xuan, J. The chloroplast genome of Carya illinoinensis: Genome structure, adaptive evolution, and phylogenetic analysis. Forests 2020, 11, 207. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wuyun, T.N.; Du, H.; Wang, D.; Cao, D. Complete chloroplast genome sequences of Eucommia ulmoides: Genome structure and evolution. Tree Genet. Genomes 2016, 12, 12. [Google Scholar] [CrossRef]
- Liu, Q.; Li, X.; Li, M.; Xu, W.; Heslop-Harrison, J.S. Comparative chloroplast genome analyses of avena: Insights into evolutionary dynamics and phylogeny. BMC Plant Biol. 2020, 20, 406. [Google Scholar] [CrossRef]
- McDonald, M.J.; Wang, W.C.; Huang, H.D.; Leu, J.Y. Clusters of nucleotide substitutions and insertion/deletion mutations are associated with repeat sequences. PLoS Biol. 2011, 9, e1000622. [Google Scholar] [CrossRef] [Green Version]
- Dugas, D.; Hernandez, D.; Koenen, E.; Schwarz, E.; Straub, S.; Hughes, C.E.; Jansen, R.K.; Nageswara-Rao, M.; Staats, M.; Trujillo, J.T.; et al. Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions, and accelerated rate of evolution in clpP. Sci. Rep. 2015, 5, 16958. [Google Scholar] [CrossRef] [Green Version]
- Drescher, A.; Stephanie, R.; Calsa, T.; Carrer, H.; Bock, R. The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. 2000, 22, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Downie, S.R.; Jansen, R.K. A comparative analysis of whole plastid genomes from the Apiales: Expansion and contraction of the inverted repeat, mitochondrial to plastid transfer of DNA, and identification of highly divergent noncoding regions. Syst. Bot. 2015, 40, 336–351. [Google Scholar] [CrossRef]
- Chumley, T.W.; Palmer, J.D.; Mower, J.P.; Fourcade, H.M.; Calie, P.J.; Boore, J.L.; Jansen, R.K. The complete chloroplast genome sequence of Pelargonium × hortorum: Organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 2006, 23, 2175–2190. [Google Scholar] [CrossRef]
- Lee, H.L.; Jansen, R.K.; Chumley, T.W.; Kim, K.J. Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping inversions. Mol. Biol. Evol. 2007, 24, 1161–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Group of Genes | Name of Gene |
|---|---|---|
| Self-replication | Ribosomal RNA | rrn4.5 3, rrn5 3, rrn16 3, rrn23 3 |
| Transfer RNA | trnY-GUA, trnW-CCA, trnV-UAC 1, trnV-GAC 3, trnT-UGU, trnT-GGU, trnS-UGA, trnS-GGA, trnS-GCU, trnR-UCU, trnR-ACG 3, trnQ-UUG, trnP-UGG, trnN-GUU 3, trnM-CAU, trnL-UAG, trnL-UAA 1, trnL-CAA 3, trnK-UUU 1, trnI-GAU 1,3, trnI-CAU 3, trnH-GUG, trnG-UCC, trnG-GCC 1, trnfM-CAU 4, trnF-GAA, trnE-UUC, trnD-GUC, trnC-GCA, RNA-UGC 1,3 | |
| Small subunit of ribosome | rps2, rps3, rps4, rps7 3, rps8, rps11, rps12 2,3, rps14, rps15, rps16 1, rps18, rps19 | |
| Large subunit of ribosome | rpl2 1,3, rpl14, rpl16 1, rpl20, rpl22, rpl23 3, rpl32, rpl33, rpl36 | |
| RNA polymerase subunits | rpoA, rpoB, rpoC1 1, rpoC2 | |
| Subunits of photosystem I | psaA, psaB, psaC, psaI, psaJ | |
| Subunits of photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT | |
| Photosynthesis | Subunits of cytochrome | petA, petB 1, petD 1, petG, petL, petN |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF 1, atpH, atpI | |
| Large subunit of RuBisCO | rbcL | |
| Subunits of NADH | ndhA 1, ndhB 1,3, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Other gene | Maturase | matK |
| Envelope membrane protein | cemA | |
| Subunit of acetyl-CoA | accD | |
| C-type cytochrome synthesis gene | ccsA | |
| Protease | clpP 2 | |
| Unknown function | Conserved open reading frames | ycf1, ycf2 3, ycf3 2, ycf4, ihbA |
| Gene | Region (bp) | Exon Ⅰ (bp) | Intron Ⅰ (bp) | Exon Ⅱ (bp) | Intron Ⅱ (bp) | Exon Ⅲ (bp) |
|---|---|---|---|---|---|---|
| atpF | LSC | 144 — | 762 | 411 — | ||
| clpP | LSC | 71 — | 847 | 292 — | 617 | 227 — |
| ndhA | SSC | 552 — | 1211 | 540 — | ||
| ndhB | IRB | 777 — | 686 | 762 — | ||
| ndhB | IRA | 775 + | 686 | 760 + | ||
| petB | LSC | 4 + | 822 | 640 + | ||
| petD | LSC | 6 + | 615 | 485 + | ||
| rpl16 | LSC | 9 — | 919 | 399 — | ||
| rpl2 | IRB | 390 — | 663 | 435 — | ||
| rpl2 | IRA | 388 + | 663 | 433 + | ||
| rpoC1 | LSC | 430 — | 843 | 1619 — | ||
| rps12 | IRB | 114 — | - | 229 + | 537 | 29 + |
| rps12 | IRA | 114 — | - | 231 — | 537 | 29 — |
| rps16 | LSC | 40 — | 894 | 230 — | ||
| trnA-UGC | IRB | 36 + | 801 | 40 + | ||
| trnA-UGC | IRA | 38 — | 801 | 42 — | ||
| trnG-GCC | LSC | 22 + | 715 | 45 + | ||
| trnI-GAU | IRB | 40 + | 950 | 33 + | ||
| trnI-GAU | IRA | 42 — | 950 | 35 — | ||
| trnK-UUU | LSC | 37 — | 2557 | 35 — | ||
| trnL-UAA | LSC | 35 + | 524 | 48 + | ||
| trnV-UAC | LSC | 38 — | 615 | 37 — | ||
| ycf3 | LSC | 126 — | 720 | 229 — | 793 | 151 — |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, J.; Li, X.; Chen, X.; Huang, X.; Jin, S. The Complete Chloroplast Genome of Carya cathayensis and Phylogenetic Analysis. Genes 2022, 13, 369. https://doi.org/10.3390/genes13020369
Shen J, Li X, Chen X, Huang X, Jin S. The Complete Chloroplast Genome of Carya cathayensis and Phylogenetic Analysis. Genes. 2022; 13(2):369. https://doi.org/10.3390/genes13020369
Chicago/Turabian StyleShen, Jianshuang, Xueqin Li, Xia Chen, Xiaoling Huang, and Songheng Jin. 2022. "The Complete Chloroplast Genome of Carya cathayensis and Phylogenetic Analysis" Genes 13, no. 2: 369. https://doi.org/10.3390/genes13020369
APA StyleShen, J., Li, X., Chen, X., Huang, X., & Jin, S. (2022). The Complete Chloroplast Genome of Carya cathayensis and Phylogenetic Analysis. Genes, 13(2), 369. https://doi.org/10.3390/genes13020369