
Integrated Analysis of RNA Binding Protein-Related lncRNA Prognostic Signature for Breast Cancer Patients



Abstract
:1. Introduction
2. Materials and Methods
2.1. Acquisition and Processing of Data
2.2. Identification of Differential Expressed RBP-Related lncRNAs in BC Tissues
2.3. Construction of the RBP-Related Prognostic Signature
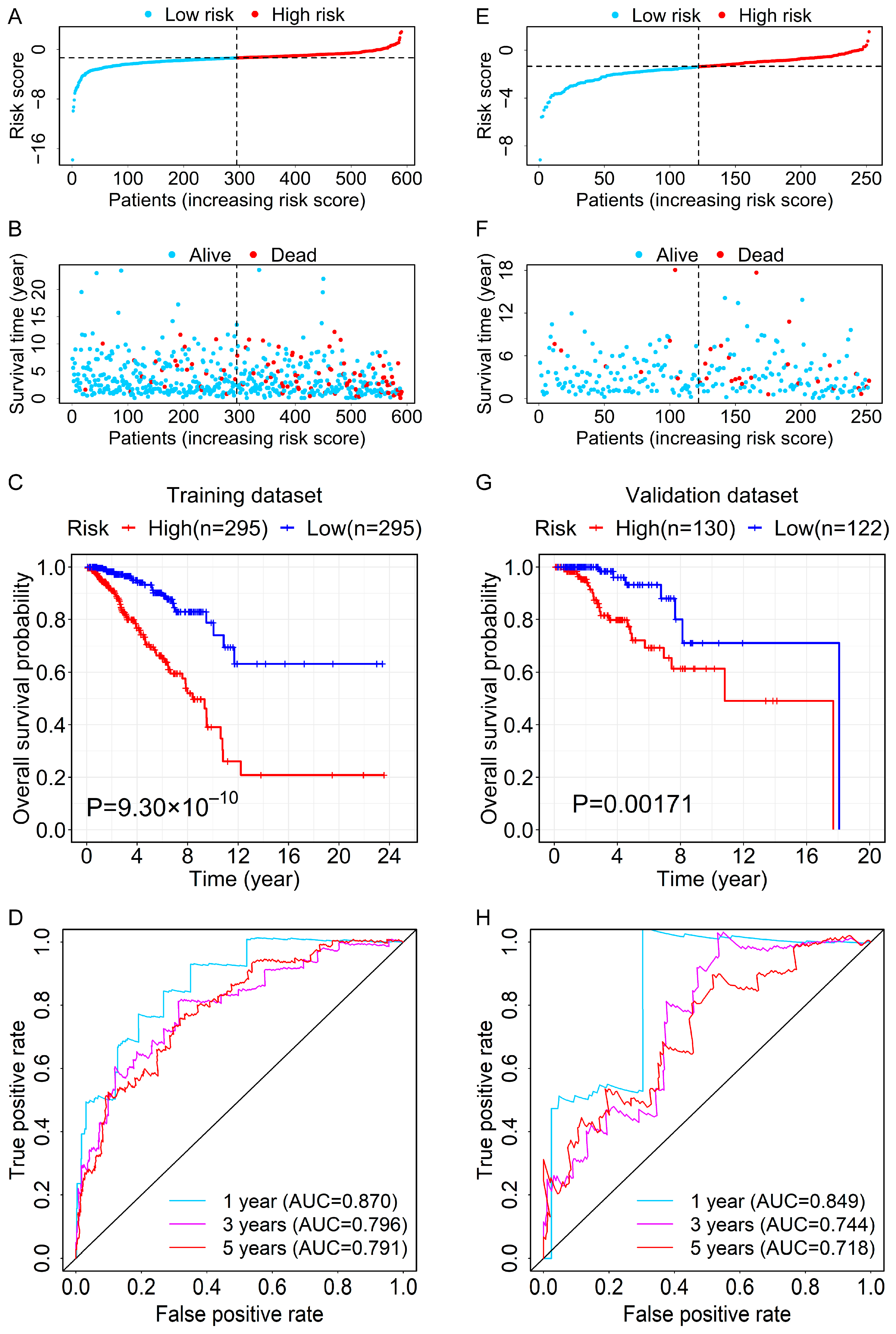
2.4. Assessment and Validation for the Prognostic Value of the RBP-Related lncRNA Signature
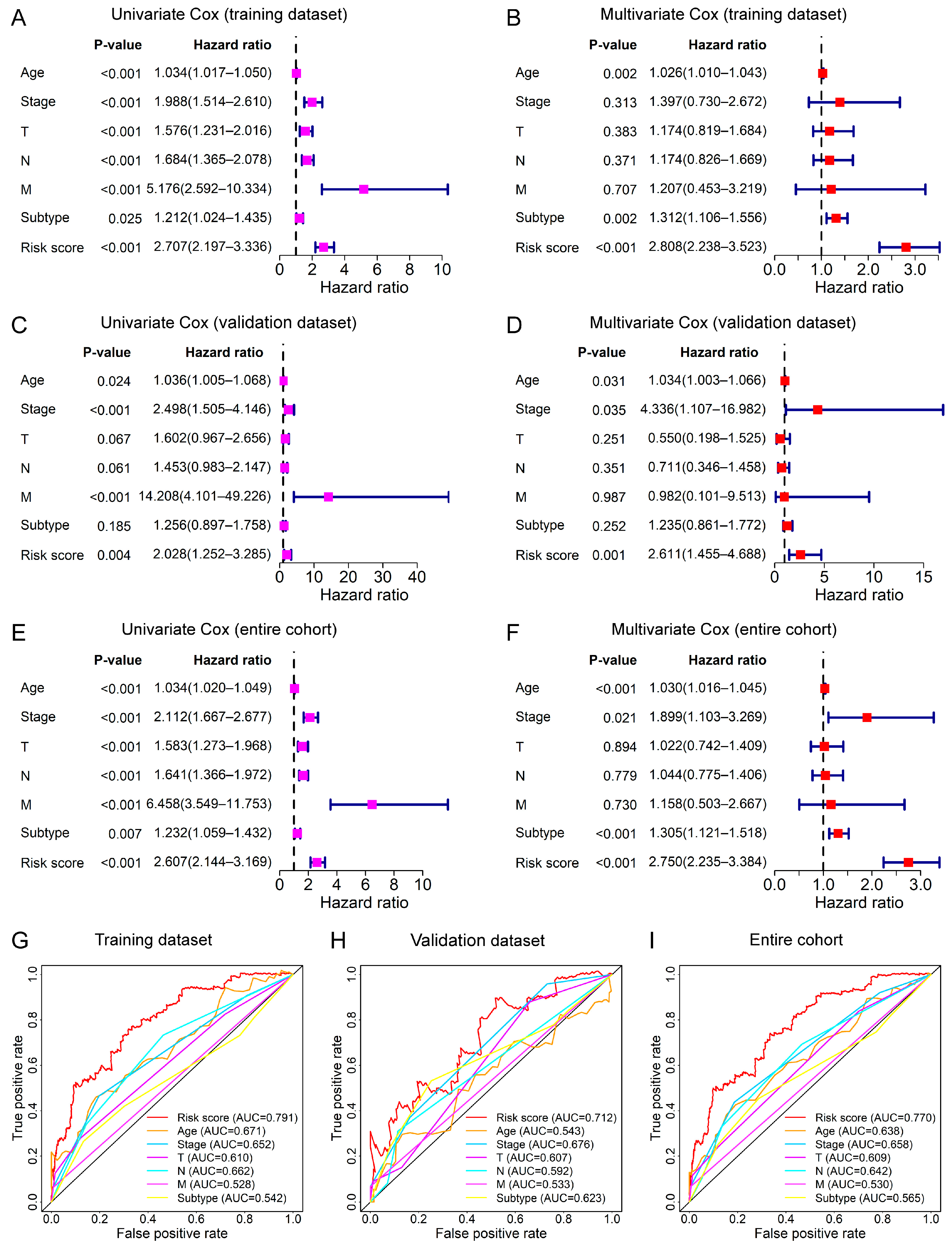
2.5. Clinical Correlation Analysis for Risk Score
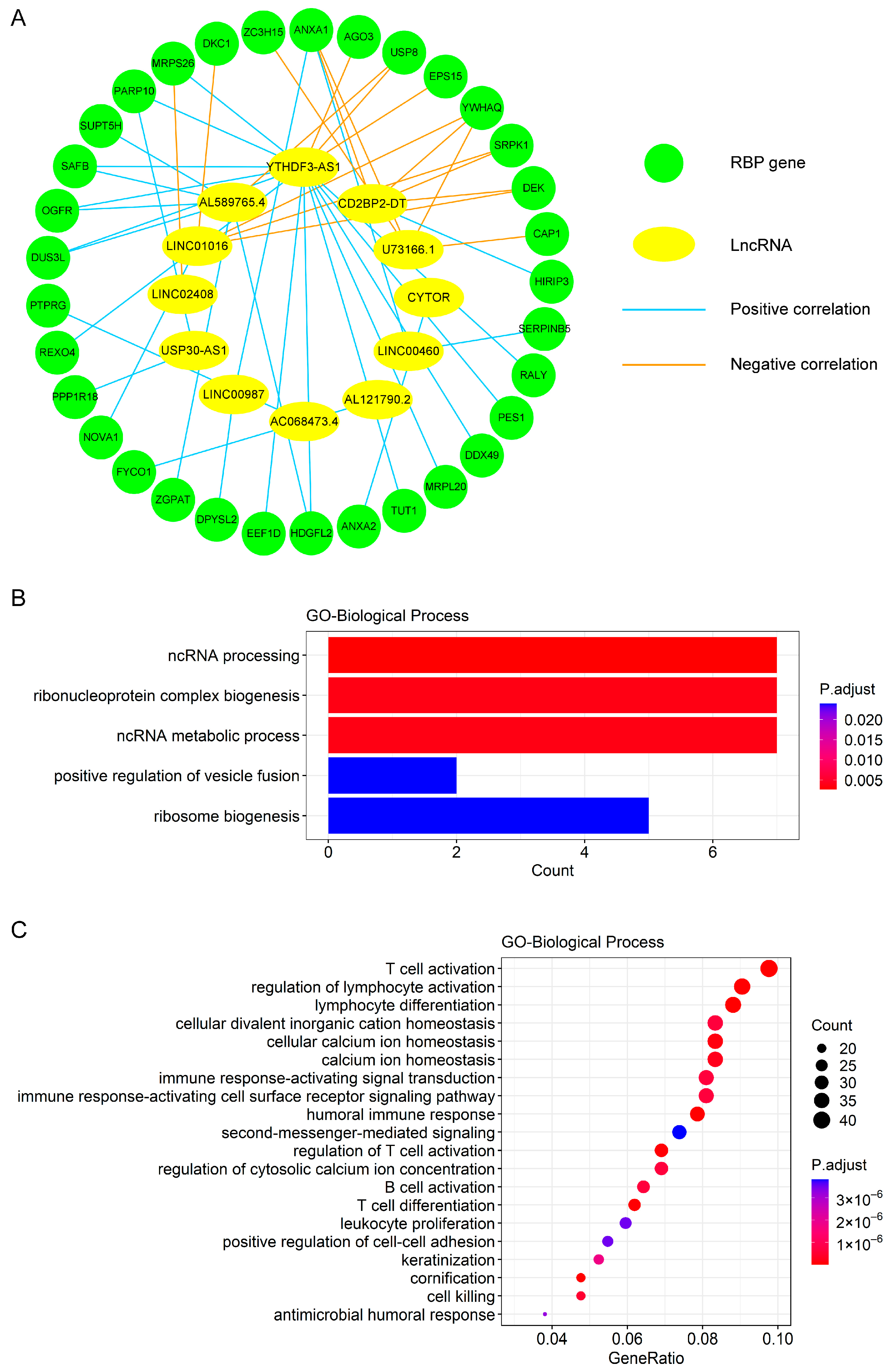
2.6. Functional Enrichment Analysis
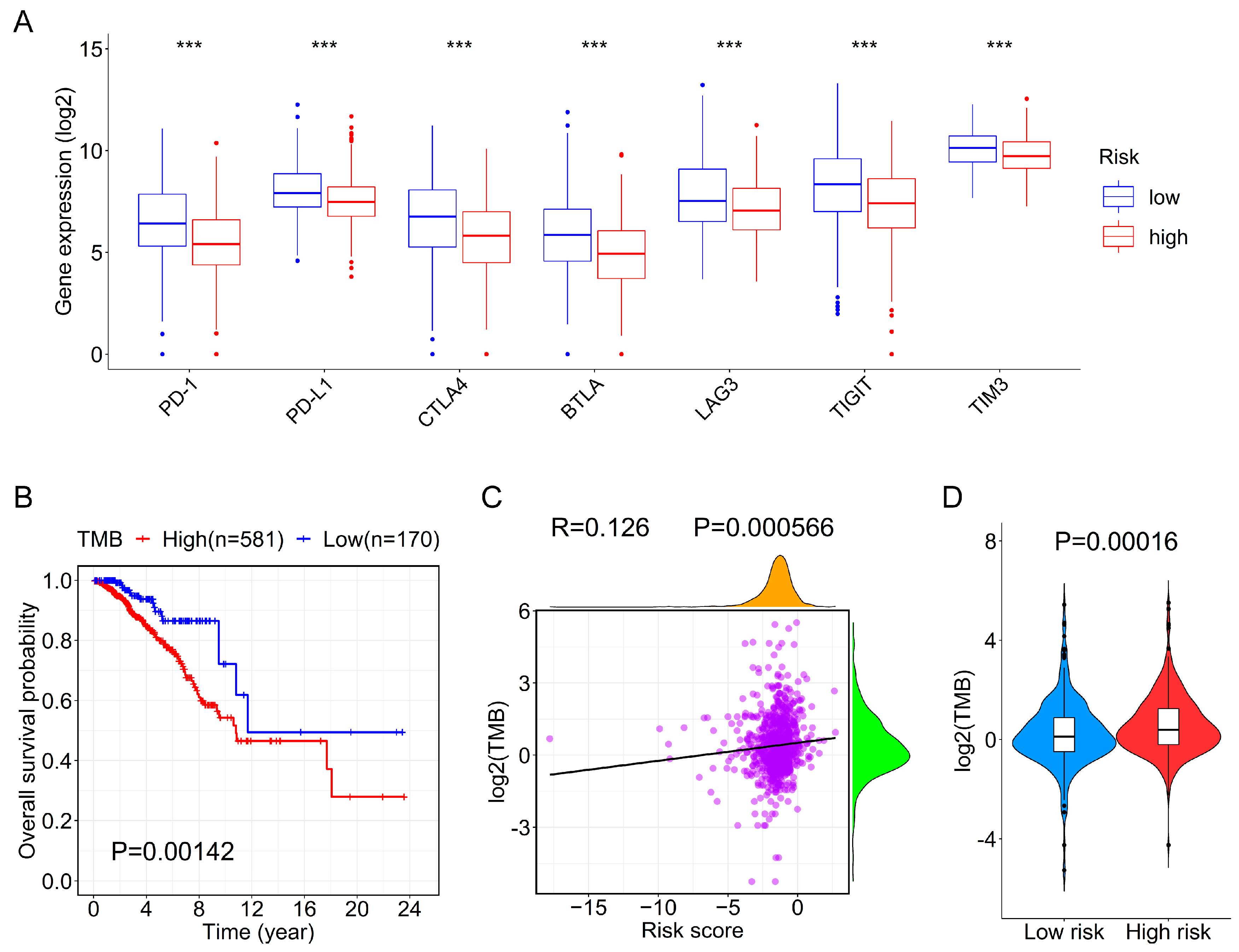
2.7. Correlation between the Risk Score and Tumor Mutational Burden
2.8. Statistical Analysis
3. Results
3.1. Identification of Differentially Expressed lncRNAs Associated with RBPs in Female BC Patients
3.2. Development of the RBP-Associated lncRNA Signature
3.3. Evaluation and Validation for the Prognostic Ability of the RBP-Related lncRNA Signature
3.4. Relationship between the Prognostic Signature and Clinical Features
3.5. Construction of Co-Expression Network and Functional Enrichment Analysis for Exploring Biological Processes Relevant to the Prognostic Signature
3.6. Association between the Prognostic Signature and Immune Checkpoint Genes or TMB
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment A Review. JAMA-J. Am. Med. Assoc. 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef]
- Pereira, B.; Billaud, M.; Almeida, R. RNA-Binding Proteins in Cancer: Old Players and New Actors. Trends Cancer 2017, 3, 506–528. [Google Scholar] [CrossRef]
- Qin, H.; Ni, H.; Liu, Y.; Yuan, Y.; Xi, T.; Li, X.; Zheng, L. RNA-binding proteins in tumor progression. J. Hematol. Oncol. 2020, 13, 90. [Google Scholar] [CrossRef]
- Yao, R.W.; Wang, Y.; Chen, L.L. Cellular functions of long noncoding RNAs. Nat. Cell Biol. 2019, 21, 542–551. [Google Scholar] [CrossRef]
- Amelio, I.; Bernassola, F.; Candi, E. Emerging roles of long non-coding RNAs in breast cancer biology and management. Semin. Cancer Biol. 2021, 72, 36–45. [Google Scholar] [CrossRef]
- Lin, S.; Zhao, M.; Lv, Y.; Mao, G.; Ding, S.; Peng, F. The lncRNA GATA3-AS1/miR-495-3p/CENPU axis predicts poor prognosis of breast cancer via the PLK1 signaling pathway. Aging 2021, 13, 13663–13679. [Google Scholar] [CrossRef]
- Zhang, Y.; Dong, X.; Wang, Y.; Wang, L.; Han, G.; Jin, L.; Fan, Y.; Xu, G.; Yuan, D.; Zheng, J.; et al. Overexpression of LncRNA BM466146 Predicts Better Prognosis of Breast Cancer. Front. Oncol. 2020, 10, 628757. [Google Scholar] [CrossRef]
- Grelet, S.; Link, L.A.; Howley, B.; Obellianne, C.; Palanisamy, V.; Gangaraju, V.K.; Diehl, J.A.; Howe, P.H. A regulated PNUTS mRNA to lncRNA splice switch mediates EMT and tumour progression. Nat. Cell Biol. 2017, 19, 1105–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, Y.; Liu, J.; Zhang, Z.; Liu, L. HuR-regulated lncRNA NEAT1 stability in tumorigenesis and progression of ovarian cancer. Cancer Med. 2016, 5, 1588–1598. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.P.; Jin, Y.P.; Wu, X.S.; Yang, Y.; Li, Y.S.; Li, H.F.; Xiang, S.S.; Song, X.L.; Jiang, L.; Zhang, Y.J.; et al. LncRNA-HGBC stabilized by HuR promotes gallbladder cancer progression by regulating miR-502-3p/SET/AKT axis. Mol. Cancer 2019, 18, 167. [Google Scholar] [CrossRef]
- Yoon, J.H.; Abdelmohsen, K.; Srikantan, S.; Yang, X.; Martindale, J.L.; De, S.; Huarte, M.; Zhan, M.; Becker, K.G.; Gorospe, M. LincRNA-p21 suppresses target mRNA translation. Mol. Cell 2012, 47, 648–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.H.; Abdelmohsen, K.; Kim, J.; Yang, X.; Martindale, J.L.; Tominaga-Yamanaka, K.; White, E.J.; Orjalo, A.V.; Rinn, J.L.; Kreft, S.G.; et al. Scaffold function of long non-coding RNA HOTAIR in protein ubiquitination. Nat. Commun. 2013, 4, 2939. [Google Scholar] [CrossRef] [PubMed]
- Hammerle, M.; Gutschner, T.; Uckelmann, H.; Ozgur, S.; Fiskin, E.; Gross, M.; Skawran, B.; Geffers, R.; Longerich, T.; Breuhahn, K.; et al. Posttranscriptional destabilization of the liver-specific long noncoding RNA HULC by the IGF2 mRNA-binding protein 1 (IGF2BP1). Hepatology 2013, 58, 1703–1712. [Google Scholar] [CrossRef]
- Tian, F.J.; He, X.Y.; Wang, J.; Li, X.; Ma, X.L.; Wu, F.; Zhang, J.; Liu, X.R.; Qin, X.L.; Zhang, Y.; et al. Elevated Tristetraprolin Impairs Trophoblast Invasion in Women with Recurrent Miscarriage by Destabilization of HOTAIR. Mol. Ther. Nucleic. Acids 2018, 12, 600–609. [Google Scholar] [CrossRef]
- Jonas, K.; Calin, G.A.; Pichler, M. RNA-Binding Proteins as Important Regulators of Long Non-Coding RNAs in Cancer. Int. J. Mol. Sci. 2020, 21, 2969. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.H.; De, S.; Srikantan, S.; Abdelmohsen, K.; Grammatikakis, I.; Kim, J.; Kim, K.M.; Noh, J.H.; White, E.J.; Martindale, J.L.; et al. PAR-CLIP analysis uncovers AUF1 impact on target RNA fate and genome integrity. Nat. Commun. 2014, 5, 5248. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, Y.B.; Kleinman, C.L.; Landry-Voyer, A.M.; Majewski, J.; Bachand, F. Polyadenylation-dependent control of long noncoding RNA expression by the poly(A)-binding protein nuclear 1. PLoS Genet. 2012, 8, e1003078. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Li, X.; Yu, L.; Wang, R.; Hua, D.; Shi, C.; Sun, C.; Luo, W.; Rao, C.; Jiang, Z.; et al. The RNA-binding protein SRSF1 is a key cell cycle regulator via stabilizing NEAT1 in glioma. J. Biochem. Cell Biol. 2019, 113, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Liu, T.; Fu, H.; Li, W.; Zheng, X. Genome-wide analysis of lncRNA stability in human. PLoS Comput. Biol. 2021, 17, e1008918. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Liu, B.; Guo, H. The role of M(6)A modification in the regulation of tumor-related lncRNAs. Mol. Ther.-Nucleic Acids 2021, 24, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.Z.; Chai, R.C.; Pang, B.; Chang, X.; An, S.Y.; Zhang, K.N.; Jiang, T.; Wang, Y.Z. METTL3 enhances the stability of MALAT1 with the assistance of HuR via m6A modification and activates NF-kappaB to promote the malignant progression of IDH-wildtype glioma. Cancer Lett. 2021, 511, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, S.; He, C.; Xue, P.; Zhang, L.; He, Z.; Zang, L.; Feng, B.; Sun, J.; Zheng, M. METTL14 suppresses proliferation and metastasis of colorectal cancer by down-regulating oncogenic long non-coding RNA XIST. Mol. Cancer 2020, 19, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Han, L.; Hu, X.; Sun, T.; Xu, D.; Li, Y.; Chen, Q.; Yao, W.; He, M.; Wang, Z.; et al. N6-methyladenosine reader IMP2 stabilizes the ZFAS1/OLA1 axis and activates the Warburg effect: Implication in colorectal cancer. J. Hematol. Oncol. 2021, 14, 188. [Google Scholar] [CrossRef]
- Estell, C.; Davidson, L.; Steketee, P.C.; Monier, A.; West, S. ZC3H4 restricts non-coding transcription in human cells. eLife 2021, 10, e67305. [Google Scholar] [CrossRef]
- Austenaa, L.M.I.; Piccolo, V.; Russo, M.; Prosperini, E.; Polletti, S.; Polizzese, D.; Ghisletti, S.; Barozzi, I.; Diaferia, G.R.; Natoli, G. A first exon termination checkpoint preferentially suppresses extragenic transcription. Nat. Struct. Mol. Biol. 2021, 28, 337–346. [Google Scholar] [CrossRef]
- Li, X.; Jin, F.; Li, Y. A novel autophagy-related lncRNA prognostic risk model for breast cancer. J. Cell. Mol. Med. 2021, 25, 4–14. [Google Scholar] [CrossRef]
- Li, Z.; Zheng, J.; Feng, Y.; Li, Y.; Liang, Y.; Liu, Y.; Wang, X.; Yang, Q. Integrated analysis identifies a novel lncRNA prognostic signature associated with aerobic glycolysis and hub pathways in breast cancer. Cancer Med. 2021, 10, 7877–7892. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Yu, X.; Jin, F. Identification and validation of stemness-related lncRNA prognostic signature for breast cancer. J. Transl. Med. 2020, 18, 331. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Peng, X.; Shen, C. Identification and validation of immune-related lncRNA prognostic signature for breast cancer. Genomics 2020, 112, 2640–2646. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Xiao, R. An emerging role of chromatin-interacting RNA-binding proteins in transcription regulation. Essays Biochem. 2020, 64, 907–918. [Google Scholar] [PubMed]
- Kuhn, M. Building predictive models in R using the caret package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Weng, Y.; Pan, Y.; Huang, Z.; Chen, X.; Hong, W.; Lin, T.; Wang, L.; Liu, W.; Qiu, S. A Novel Prognostic Signature Based on Metabolism-Related Genes to Predict Survival and Guide Personalized Treatment for Head and Neck Squamous Carcinoma. Front. Oncol. 2021, 11, 685026. [Google Scholar] [CrossRef]
- Therneau, T. A Package for Survival Analysis in S. R Package Version 2.37-7; Springer: New York, NY, USA, 2014. [Google Scholar]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Lu, Q.; Ren, S.; Lu, M.; Zhang, Y.; Zhu, D.; Zhang, X.; Li, T. Computational prediction of associations between long non-coding RNAs and proteins. BMC Genom. 2013, 14, 651. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Li, L.; Zhang, H.; Zhao, Y.; Zhang, H.; Wu, S.; Xu, B. A risk model developed based on tumor microenvironment predicts overall survival and associates with tumor immunity of patients with lung adenocarcinoma. Oncogene 2021, 40, 4413–4424. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Pareja, F.G. New Advances in Molecular Breast Cancer Pathology. Semin. Cancer Biol. 2021, 72, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Zhao, L.; Zhang, H.; Feng, X.; Xu, H.; Hao, J.; Wang, S.; Yang, Q.; Zou, L.; Su, X.; et al. Loss of TDP43 inhibits progression of triple-negative breast cancer in coordination with SRSF3. Proc. Natl. Acad. Sci. USA 2018, 115, E3426–E3435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, L.; Fu, L.; Yuan, Y.; Dai, H.; Zhu, T.; Zhou, Y.; Yuan, F. Integrated Bioinformatics Analysis the Function of RNA Binding Proteins (RBPs) and Their Prognostic Value in Breast Cancer. Front. Pharmacol. 2019, 10, 140. [Google Scholar] [CrossRef]
- Chen, F.; Wang, Q.; Yu, X.; Yang, N.; Wang, Y.; Zeng, Y.; Zheng, Z.; Zhou, F.; Zhou, Y. MCPIP1-mediated NFIC alternative splicing inhibits proliferation of triple-negative breast cancer via cyclin D1-Rb-E2F1 axis. Cell Death Dis. 2021, 12, 370. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, H.; Li, X.; Liu, Q.; Zhao, Y.; Li, L.; Xu, B.; Hou, Y.; Jin, W. Identification of a Three-RNA Binding Proteins (RBPs) Signature Predicting Prognosis for Breast Cancer. Front. Oncol. 2021, 11, 663556. [Google Scholar] [CrossRef]
- Jung, S.Y.; Papp, J.C.; Sobel, E.M.; Yu, H.; Zhang, Z.F. Breast Cancer Risk and Insulin Resistance: Post Genome-Wide Gene-Environment Interaction Study Using a Random Survival Forest. Cancer Res. 2019, 79, 2784–2794. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Yang, L.; Chong, Q.Y.; Yan, H.; Zhang, W.; Qian, W.; Tan, S.; Wu, Z.; Lobie, P.E.; Zhu, T. Long noncoding RNA Linc00460 promotes breast cancer progression by regulating the miR-489-5p/FGF7/AKT axis. Cancer Manag. Res. 2019, 11, 5983–6001. [Google Scholar] [CrossRef] [Green Version]
- Van Grembergen, O.; Bizet, M.; de Bony, E.J.; Calonne, E.; Putmans, P.; Brohee, S.; Olsen, C.; Guo, M.; Bontempi, G.; Sotiriou, C.; et al. Portraying breast cancers with long noncoding RNAs. Sci. Adv. 2016, 2, e1600220. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wang, X.; Zhou, L.; Jiang, M.; Zhong, G.; Xu, S.; Zhang, M.; Zhang, Y.; Liang, X.; Zhang, L.; et al. A Positive Feedback Loop of Long Noncoding RNA LINC00152 and KLF5 Facilitates Breast Cancer Growth. Front. Oncol. 2021, 11, 619915. [Google Scholar] [CrossRef]
- Li, H.; Gao, C.; Liu, L.; Zhuang, J.; Yang, J.; Liu, C.; Zhou, C.; Feng, F.; Sun, C. 7-lncRNA Assessment Model for Monitoring and Prognosis of Breast Cancer Patients: Based on Cox Regression and Co-expression Analysis. Front. Oncol. 2019, 9, 1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Yi, X.; Ji, Z.; Yao, M.; Yang, Y.; Song, W.; Huang, M. Development of a Prognostic Model Based on the Identification of EMT-Related lncRNAs in Triple-Negative Breast Cancer. J. Oncol. 2021, 2021, 9219961. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Ping, L.; Du, T.; Liang, G.; Huang, Y.; Li, Z.; Deng, R.; Tang, J. A Ferroptosis-Related lncRNAs Signature Predicts Prognosis and Immune Microenvironment for Breast Cancer. Front. Mol. Biosci. 2021, 8, 678877. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shan, B.; Lin, L.; Dong, J.; Sun, Q.; Zhou, Q.; Chen, J.; Han, X. Dissecting the Role of N6-Methylandenosine-Related Long Non-coding RNAs Signature in Prognosis and Immune Microenvironment of Breast Cancer. Front. Cell Dev. Biol. 2021, 9, 711859. [Google Scholar] [CrossRef]
- Cheng, L.; Li, J.; Han, Y.; Lin, J.; Niu, C.; Zhou, Z.; Yuan, B.; Huang, K.; Li, J.; Jiang, K.; et al. PES1 promotes breast cancer by differentially regulating ERalpha and ERbeta. J. Clin. Investig. 2012, 122, 2857–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobral-Leite, M.; Wesseling, J.; Smit, V.T.; Nevanlinna, H.; van Miltenburg, M.H.; Sanders, J.; Hofland, I.; Blows, F.M.; Coulson, P.; Patrycja, G.; et al. Annexin A1 expression in a pooled breast cancer series: Association with tumor subtypes and prognosis. BMC Med. 2015, 13, 156. [Google Scholar] [CrossRef] [Green Version]
- Bai, F.; Zhang, P.; Fu, Y.; Chen, H.; Zhang, M.; Huang, Q.; Li, D.; Li, B.; Wu, K. Targeting ANXA1 abrogates Treg-mediated immune suppression in triple-negative breast cancer. J. Immunother. Cancer 2020, 8, e000169. [Google Scholar] [CrossRef] [PubMed]
- van Roosmalen, W.; Le Devedec, S.E.; Golani, O.; Smid, M.; Pulyakhina, I.; Timmermans, A.M.; Look, M.P.; Zi, D.; Pont, C.; de Graauw, M.; et al. Tumor cell migration screen identifies SRPK1 as breast cancer metastasis determinant. J. Clin. Investig. 2015, 125, 1648–1664. [Google Scholar] [CrossRef] [Green Version]
- Elsharawy, K.A.; Mohammed, O.J.; Aleskandarany, M.A.; Hyder, A.; El-Gammal, H.L.; Abou-Dobara, M.I.; Green, A.R.; Dalton, L.W.; Rakha, E.A. The nucleolar-related protein Dyskerin pseudouridine synthase 1 (DKC1) predicts poor prognosis in breast cancer. Br. J. Cancer 2020, 123, 1543–1552. [Google Scholar] [CrossRef]
- Bergqvist, M.; Elebro, K.; Sandsveden, M.; Borgquist, S.; Rosendahl, A.H. Effects of tumor-specific CAP1 expression and body constitution on clinical outcomes in patients with early breast cancer. Breast Cancer Res. 2020, 22, 67. [Google Scholar] [CrossRef]
- Rivers, C.; Idris, J.; Scott, H.; Rogers, M.; Lee, Y.B.; Gaunt, J.; Phylactou, L.; Curk, T.; Campbell, C.; Ule, J.; et al. iCLIP identifies novel roles for SAFB1 in regulating RNA processing and neuronal function. BMC Biol. 2015, 13, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzoi, M.A.; Romano, E.; Piccart, M. Immunotherapy for early breast cancer: Too soon, too superficial, or just right? Ann. Oncol. 2021, 32, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Gaynor, N.; Crown, J.; Collins, D.M. Immune checkpoint inhibitors: Key trials and an emerging role in breast cancer. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Deng, Z.M.; Hu, W.; Dai, F.F.; Yuan, M.Q.; Hu, M.; Cheng, Y.X. Immune-Related Genes to Construct a Novel Prognostic Model of Breast Cancer: A Chemosensitivity-Based Study. Front. Immunol. 2021, 12, 734745. [Google Scholar] [CrossRef] [PubMed]
- Shum, B.; Larkin, J.; Turajlic, S. Predictive biomarkers for response to immune checkpoint inhibition. Semin. Cancer Biol. 2021, in press. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Ensembl ID | Genomic Location | Coefficient |
|---|---|---|---|
| LINC02408 | ENSG00000203585 | Chr12:67,443,105–67,590,771 | 0.007062056 |
| AL121790.2 | ENSG00000259087 | Chr14:37,556,158–37,567,095 | 0.00683141 |
| AL589765.4 | ENSG00000249602 | Chr1:151,763,384–151,769,501 | 0.003727625 |
| LINC00460 | ENSG00000233532 | Chr13:106,374,477–106,384,315 | 0.001867941 |
| YTHDF3-AS1 | ENSG00000270673 | Chr8:63,167,725–63,168,442 | 0.00146521 |
| CYTOR | ENSG00000222041 | Chr2:87,454,781–87,636,740 | 0.000252682 |
| LINC01016 | ENSG00000249346 | Chr6:33,867,506–33,896,914 | −0.000429308 |
| CD2BP2-DT | ENSG00000260219 | Chr16:30,354,665–30,357,116 | −0.001513116 |
| LINC00987 | ENSG00000237248 | Chr12:9,240,003–9,257,960 | −0.003223765 |
| U73166.1 | ENSG00000230454 | Chr3:50,260,303–50,263,358 | −0.004631155 |
| USP30-AS1 | ENSG00000256262 | Chr12:109,052,349–109,053,984 | −0.015207547 |
| AC068473.4 | ENSG00000267409 | Chr18:79,610,747–79,612,303 | −0.048280801 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, S.; Xie, J.; Zhou, Y.; Liu, H.; Wang, Y.; Li, Z. Integrated Analysis of RNA Binding Protein-Related lncRNA Prognostic Signature for Breast Cancer Patients. Genes 2022, 13, 345. https://doi.org/10.3390/genes13020345
Xu S, Xie J, Zhou Y, Liu H, Wang Y, Li Z. Integrated Analysis of RNA Binding Protein-Related lncRNA Prognostic Signature for Breast Cancer Patients. Genes. 2022; 13(2):345. https://doi.org/10.3390/genes13020345
Chicago/Turabian StyleXu, Shaohua, Jiahui Xie, Yanjie Zhou, Hui Liu, Yirong Wang, and Zhaoyong Li. 2022. "Integrated Analysis of RNA Binding Protein-Related lncRNA Prognostic Signature for Breast Cancer Patients" Genes 13, no. 2: 345. https://doi.org/10.3390/genes13020345
APA StyleXu, S., Xie, J., Zhou, Y., Liu, H., Wang, Y., & Li, Z. (2022). Integrated Analysis of RNA Binding Protein-Related lncRNA Prognostic Signature for Breast Cancer Patients. Genes, 13(2), 345. https://doi.org/10.3390/genes13020345