
A National French Consensus on Gene List for the Diagnosis of Charcot–Marie–Tooth Disease and Related Disorders Using Next-Generation Sequencing

 , , ,
, , ,
Abstract
:
1. Introduction
1.1. CMT Is a Heterogeneous Genetic Disease
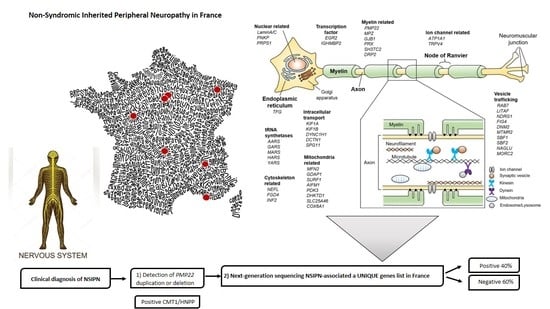
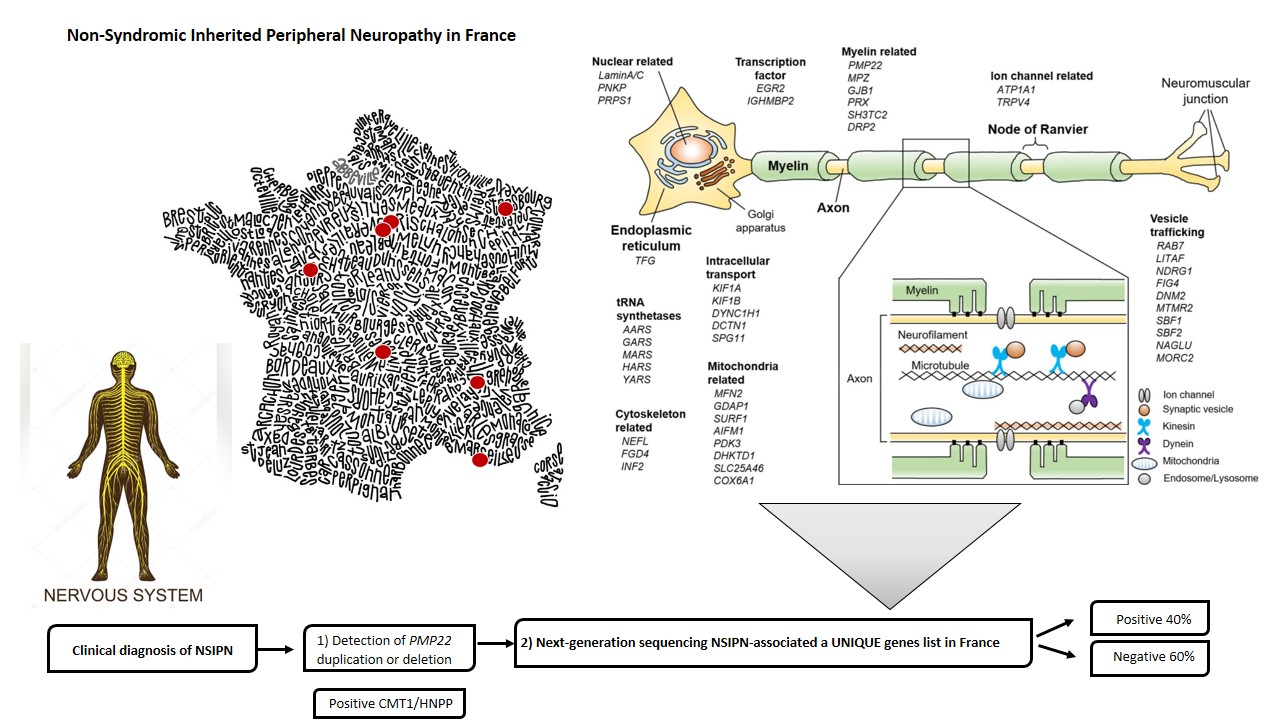
1.2. Molecular Diagnosis Is Positive around 40% of Cases
1.3. Next Generation Sequencing Technologies Increase the Rate of Molecular Diagnosis
2. Materials and Methods
3. Results and Discussion
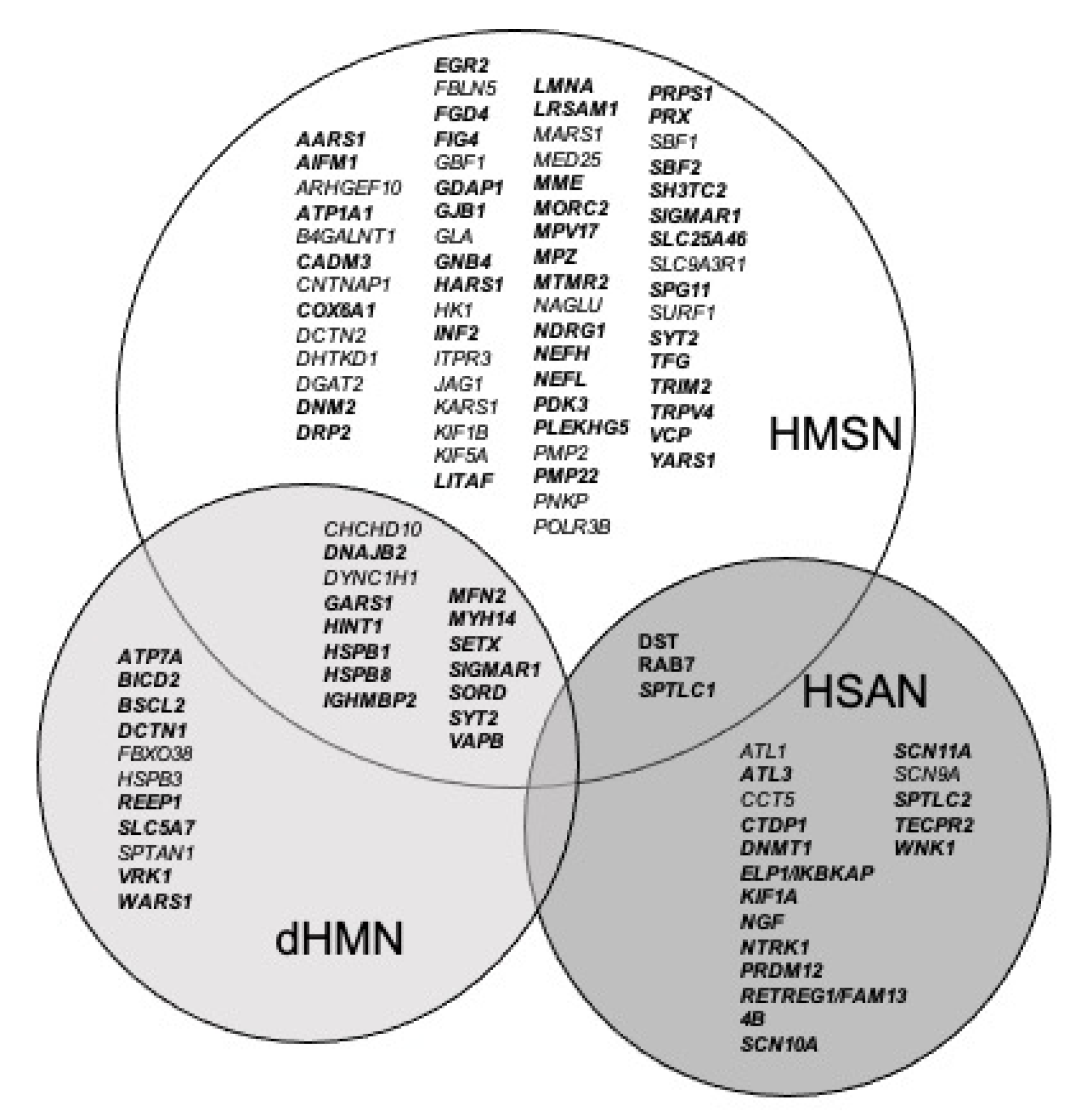
3.1. The FILNEMUS Consortium Implemented a Unique Gene Panel for NSIPN
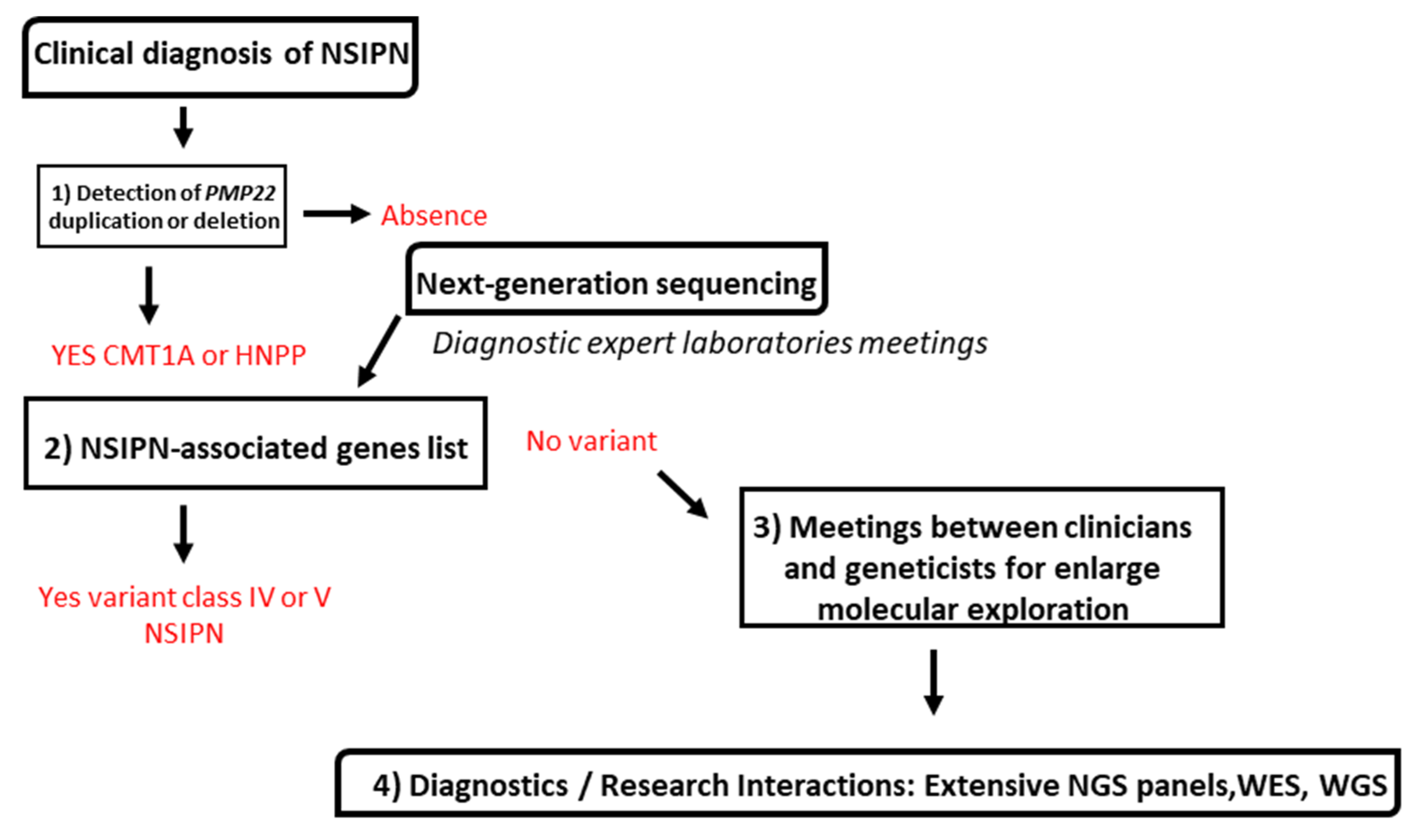
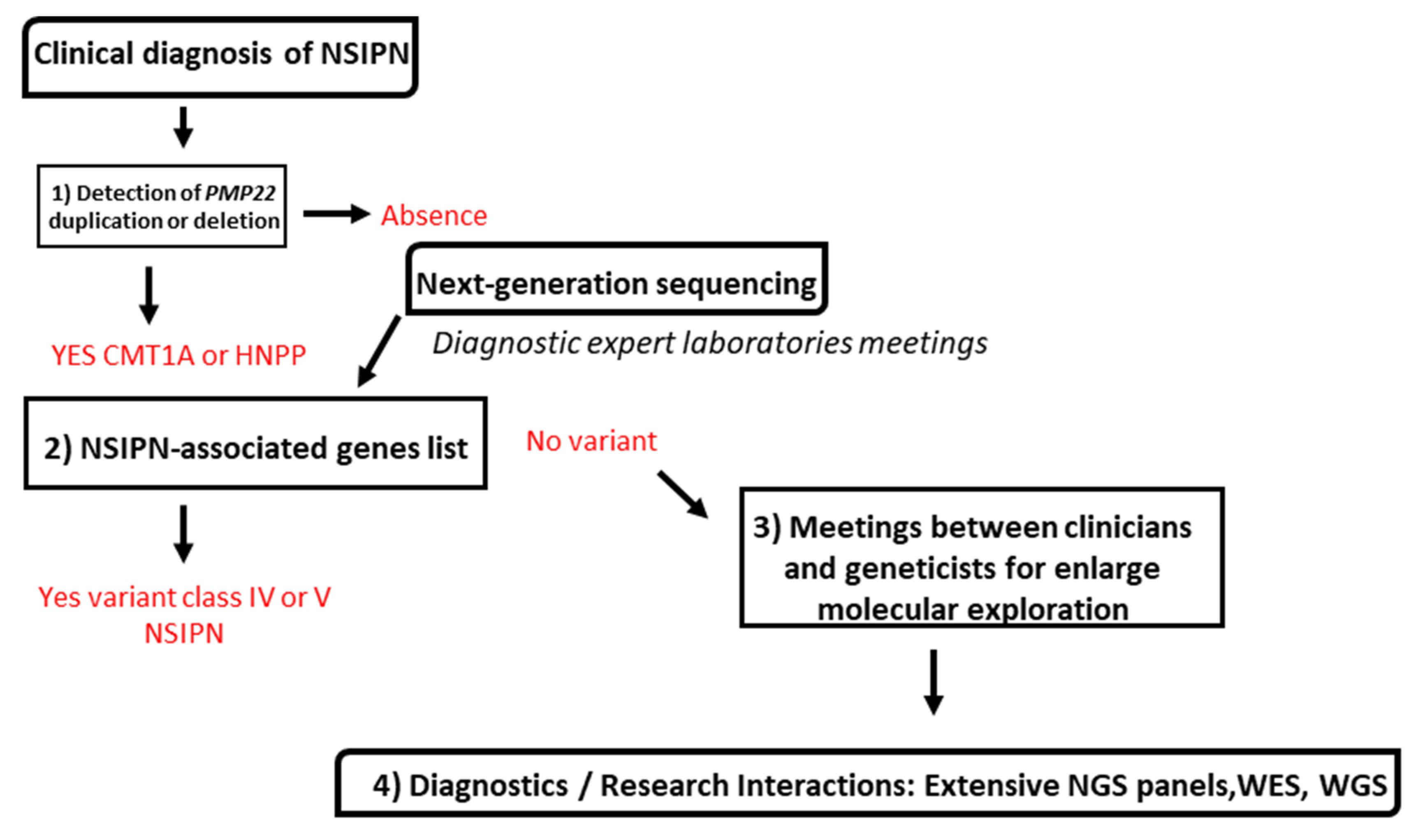
3.2. Molecular Strategy Is Based on Four Steps
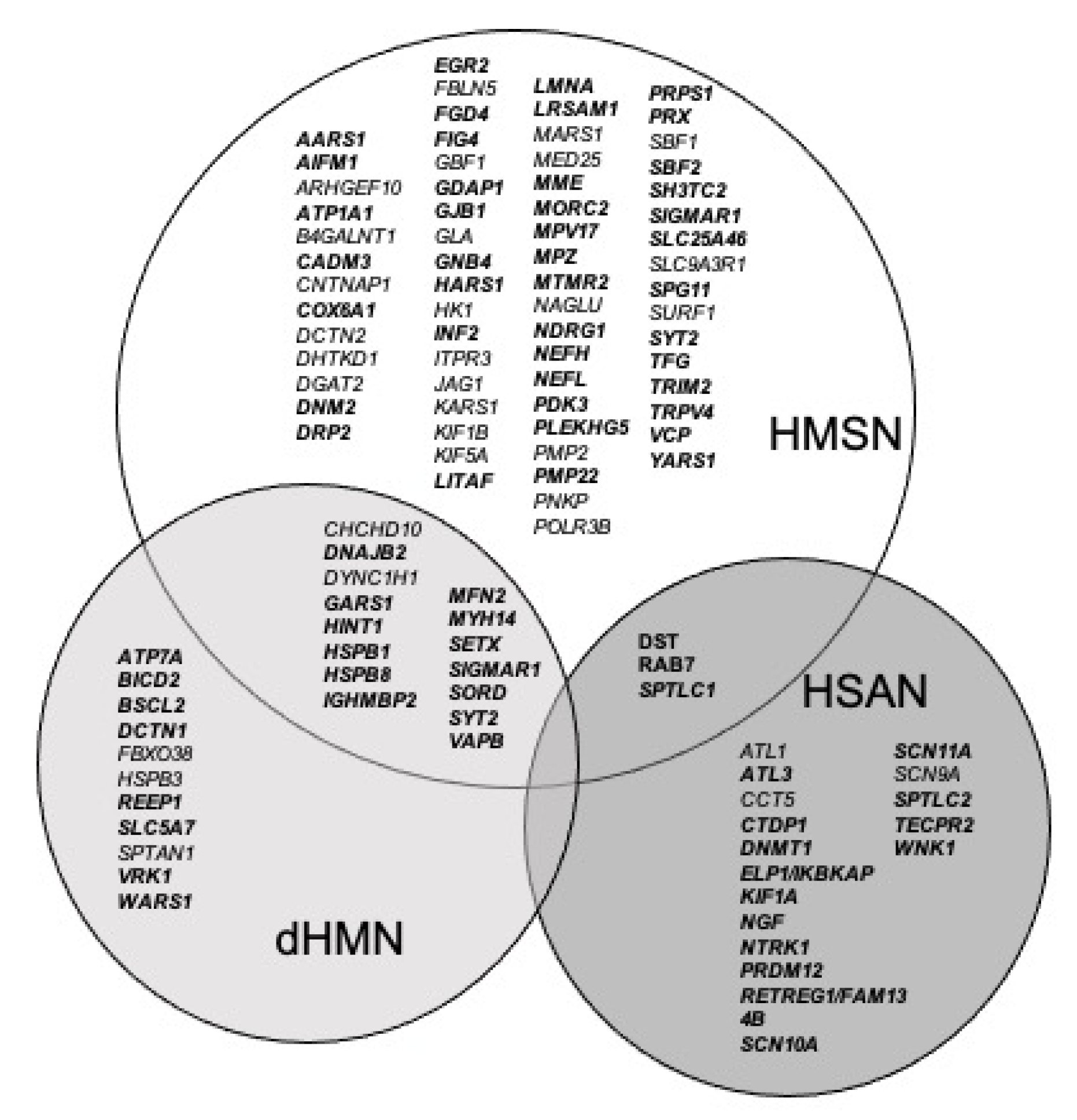
3.3. Genes Are Classified Depending on Literature Evidence Based on Strande Publication and ClinGen Evaluation
3.4. This Classification Allows Stratification of Variant Analysis
4. Conclusions
How Does This Consortium Helps Molecular Diagnosis in NSIPN?
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NGS | Next Generation Sequencing |
| CMT | Charcot–Marie–Tooth disease |
| HMSN | Hereditary Motor and Sensory Neuropathies |
| dHMN | Distal Hereditary Motor Neuropathies |
| HSAN | Hereditary Motor Neuropathies |
| DNA | Deoxyribonucleic Acid |
| FILNEMUS | Filière Nationale des Maladies Rares Neuromusculaires |
| PMP22 | peripheral myelin protein 22 |
| MPZ | Myelin Protein Zero |
| PNKP | Polynucleotide Kinase Phosphatase |
| WES | Whole Exome Sequencing |
| WGS | Whole Genome Sequencing |
| MLPA | Multiplex-ligation-dependent-probe-amplification |
| DYNC1H1 | Dynein Cytoplasmic 1 Heavy Chain 1 |
| GARS | Glycyl-TRNA Synthetase 1 |
| HSPB | Heat Shock Protein Family |
| IGHMBP2 | Immunoglobulin Mu DNA Binding Protein |
| MFN2 | Mitofusin 2 |
| PLEKHG5 | Pleckstrin Homology And RhoGEF Domain Containing G5 |
| SPG11 | Spastic Paraplegia 11 |
| VUS | Variant of Uncertain Significance |
References
- Skre, H. Genetic and Clinical Aspects of Charcot–Marie–Tooth’s Disease. Clin. Genet. 1974, 6, 98–118. [Google Scholar] [CrossRef] [PubMed]
- Pisciotta, C.; Shy, M.E. Neuropathy. Handb. Clin. Neurol. 2018, 148, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Bonne, G.; Rivier, F.; Hamroun, D. The 2019 Version of the Gene Table of Neuromuscular Disorders (Nuclear Genome). Neuromuscul. Disord. 2018, 28, 1031–1063. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Bruno, G.; Fratta, M.; Colavito, D.; Casertano, S.; Sampaolo, S.; Oliva, M.; Puoti, G. Expanding the Spectrum of SPTLC1-Related Disorders beyond Hereditary Sensory and Autonomic Neuropathies: A Novel Case of the Distinct “S331 Syndrome”. J. Peripher. Nerv. Syst. 2020, 25, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Bacquet, J.; Stojkovic, T.; Boyer, A.; Martini, N.; Audic, F.; Chabrol, B.; Salort-Campana, E.; Delmont, E.; Desvignes, J.-P.; Verschueren, A.; et al. Molecular Diagnosis of Inherited Peripheral Neuropathies by Targeted Next-Generation Sequencing: Molecular Spectrum Delineation. BMJ Open 2018, 8, e021632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartley, T.; Wagner, J.D.; Warman-Chardon, J.; Tétreault, M.; Brady, L.; Baker, S.; Tarnopolsky, M.; Bourque, P.R.; Parboosingh, J.S.; Smith, C.; et al. Whole-Exome Sequencing Is a Valuable Diagnostic Tool for Inherited Peripheral Neuropathies: Outcomes from a Cohort of 50 Families. Clin. Genet. 2018, 93, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Gonzaga-Jauregui, C.; Harel, T.; Gambin, T.; Kousi, M.; Griffin, L.B.; Francescatto, L.; Ozes, B.; Karaca, E.; Jhangiani, S.N.; Bainbridge, M.N.; et al. Exome Sequence Analysis Suggests That Genetic Burden Contributes to Phenotypic Variability and Complex Neuropathy. Cell Rep. 2015, 12, 1169–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohrn, M.F.; Glöckle, N.; Mulahasanovic, L.; Heller, C.; Mohr, J.; Bauer, C.; Riesch, E.; Becker, A.; Battke, F.; Hörtnagel, K.; et al. Frequent Genes in Rare Diseases: Panel-Based next Generation Sequencing to Disclose Causal Mutations in Hereditary Neuropathies. J. Neurochem. 2017, 143, 507–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padilha, J.P.D.; Brasil, C.S.; Hoefel, A.M.L.; Winckler, P.B.; Donis, K.C.; Brusius-Facchin, A.C.; Saute, J.A.M. Diagnostic Yield of Targeted Sequential and Massive Panel Approaches for Inherited Neuropathies. Clin. Genet. 2020, 98, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh, S.; Vazehan, R.; Beheshtian, M.; Sadeghinia, F.; Fattahi, Z.; Mohseni, M.; Arzhangi, S.; Nafissi, S.; Kariminejad, A.; Najmabadi, H.; et al. Molecular Diagnosis of Hereditary Neuropathies by Whole Exome Sequencing and Expanding the Phenotype Spectrum. Arch. Iran. Med. 2020, 23, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Cortese, A.; Wilcox, J.E.; Polke, J.M.; Poh, R.; Skorupinska, M.; Rossor, A.M.; Laura, M.; Tomaselli, P.J.; Houlden, H.; Shy, M.E.; et al. Targeted Next-Generation Sequencing Panels in the Diagnosis of Charcot–Marie–Tooth Disease. Neurology 2020, 94, e51–e61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pipis, M.; Rossor, A.M.; Laura, M.; Reilly, M.M. Next-Generation Sequencing in Charcot–Marie–Tooth Disease: Opportunities and Challenges. Nat. Rev. Neurol. 2019, 15, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Krahn, M.; Biancalana, V.; Cerino, M.; Perrin, A.; Michel-Calemard, L.; Nectoux, J.; Leturcq, F.; Bouchet-Séraphin, C.; Acquaviva-Bourdain, C.; Campana-Salort, E.; et al. A National French Consensus on Gene Lists for the Diagnosis of Myopathies Using Next-Generation Sequencing. Eur. J. Hum. Genet. 2019, 27, 349–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benarroch, L.; Bonne, G.; Rivier, F.; Hamroun, D. The 2020 Version of the Gene Table of Neuromuscular Disorders (Nuclear Genome). Neuromuscul. Disord. 2019, 29, 980–1018. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strande, N.T.; Riggs, E.R.; Buchanan, A.H.; Ceyhan-Birsoy, O.; DiStefano, M.; Dwight, S.S.; Goldstein, J.; Ghosh, R.; Seifert, B.A.; Sneddon, T.P.; et al. Evaluating the Clinical Validity of Gene-Disease Associations: An Evidence-Based Framework Developed by the Clinical Genome Resource. Am. J. Hum. Genet. 2017, 100, 895–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossor, A.M.; Carr, A.S.; Devine, H.; Chandrashekar, H.; Pelayo-Negro, A.L.; Pareyson, D.; Shy, M.E.; Scherer, S.S.; Reilly, M.M. Peripheral Neuropathy in Complex Inherited Diseases: An Approach to Diagnosis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 846–863. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Subgroup | Definitive Genes n (%) | Limited Genes n (%) | Total Genes n |
|---|---|---|---|
| CMT or HMSN | 55 (68%) | 26 (32%) | 81 |
| dHMN | 21(81%) | 5(19%) | 26 |
| HSAN | 17(85%) | 3(15%) | 20 |
| NSIPN | 93 (73%) | 34 (27%) | 127 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benquey, T.; Pion, E.; Cossée, M.; Krahn, M.; Stojkovic, T.; Perrin, A.; Cerino, M.; Molon, A.; Lia, A.-S.; Magdelaine, C.; et al. A National French Consensus on Gene List for the Diagnosis of Charcot–Marie–Tooth Disease and Related Disorders Using Next-Generation Sequencing. Genes 2022, 13, 318. https://doi.org/10.3390/genes13020318
Benquey T, Pion E, Cossée M, Krahn M, Stojkovic T, Perrin A, Cerino M, Molon A, Lia A-S, Magdelaine C, et al. A National French Consensus on Gene List for the Diagnosis of Charcot–Marie–Tooth Disease and Related Disorders Using Next-Generation Sequencing. Genes. 2022; 13(2):318. https://doi.org/10.3390/genes13020318
Chicago/Turabian StyleBenquey, Thibaut, Emmanuelle Pion, Mireille Cossée, Martin Krahn, Tanya Stojkovic, Aurélien Perrin, Mathieu Cerino, Annamaria Molon, Anne-Sophie Lia, Corinne Magdelaine, and et al. 2022. "A National French Consensus on Gene List for the Diagnosis of Charcot–Marie–Tooth Disease and Related Disorders Using Next-Generation Sequencing" Genes 13, no. 2: 318. https://doi.org/10.3390/genes13020318
APA StyleBenquey, T., Pion, E., Cossée, M., Krahn, M., Stojkovic, T., Perrin, A., Cerino, M., Molon, A., Lia, A.-S., Magdelaine, C., Francou, B., Guiochon-Mantel, A., Malinge, M.-C., Leguern, E., Lévy, N., Attarian, S., Latour, P., & Bonello-Palot, N. (2022). A National French Consensus on Gene List for the Diagnosis of Charcot–Marie–Tooth Disease and Related Disorders Using Next-Generation Sequencing. Genes, 13(2), 318. https://doi.org/10.3390/genes13020318