
Therapeutic Metabolic Reprograming Using microRNAs: From Cancer to HIV Infection


Abstract
:1. Energy Metabolism as a microRNA-Regulated Process
1.1. An Overview of Energy Metabolism
1.2. Proliferating Cells Favor Aerobic Glycolysis and Glutaminolysis
1.3. Energy Metabolism Is Tightly Linked to Biosynthesis
1.4. Metabolism Is Highly Regulated by Signalling Pathways and miRNAs
2. Metabolic Adaptations of Cancer Cells
2.1. Cancer Cell Metabolic Strategies Sustain High Proliferation in Nutrient and Oxygen-Poor Environments
2.2. LDHA Is a Critical Player in Tumor Metabolic Reprograming and Suppression of Immunosurveillance
3. miRNAs in the Therapeutic Modulation of Cancer Metabolism
3.1. miRNAs Regulating Metabolic Pathways in Cancer
3.2. Therapeutic Modulation of Metabolism in Cancer: The Power of miRNAs
3.3. miRNA Control of the TME and Immunosupression
4. Metabolism and microRNAs in the Immune System
4.1. Quick Primer on T Cell Immunometabolism
4.2. The Susceptibility of CD4 T Cells to HIV Infection Is Strongly Linked to Their Metabolism
4.3. Host Cell Metabolism Influences Virion Production, Infectivity, and Latent Virus Reactivation
5. Control of HIV-1 Replication and Latency by “Metabolic” miRNAs: A Potential Therapeutic Approach?
5.1. microRNAs Are Critical Regulators of HIV Infection
5.2. microRNA-Dependent Regulation of Metabolic Pathways in the Context of Viral Infection
5.3. Interactions between miRNAs and Metabolic Pathways Relevant for HIV Replication
6. Final Remarks
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lau, N.C.; Lim, L.P.; Weinstein, E.G.; Bartel, D.P. An Abundant Class of Tiny RNAs with Probable Regulatory Roles in Caenorhabditis elegans. Science 2001, 294, 858–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, R.C.; Ambros, V. An Extensive Class of Small RNAs in Caenorhabditis elegans. Science 2001, 294, 862–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of Novel Genes Coding for Small Expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fromm, B.; Domanska, D.; Høye, E.; Ovchinnikov, V.; Kang, W.; Aparicio-Puerta, E.; Johansen, M.; Flatmark, K.; Mathelier, A.; Hovig, E.; et al. MirGeneDB 2.0: The Metazoan MicroRNA Complement. Nucleic Acids Res. 2020, 48, D132–D141. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From MicroRNA Sequences to Function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Halushka, M.K.; Fromm, B.; Peterson, K.J.; McCall, M.N. Big Strides in Cellular MicroRNA Expression. Trends Genet 2018, 34, 165–167. [Google Scholar] [CrossRef]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of MicroRNA Biogenesis and Its Crosstalk with Other Cellular Pathways. Nat. Rev. Mol. Cell Biol. 2019, 20, 5–20. [Google Scholar] [CrossRef]
- Cheloufi, S.; Dos Santos, C.O.; Chong, M.M.W.; Hannon, G.J. A Dicer-Independent MiRNA Biogenesis Pathway That Requires Ago Catalysis. Nature 2010, 465, 584–589. [Google Scholar] [CrossRef] [Green Version]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional Regulation of the Heterochronic Gene Lin-14 by Lin-4 Mediates Temporal Pattern Formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans Heterochronic Gene Lin-4 Encodes Small RNAs with Antisense Complementarity to Lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Chen, X.; Liang, H.; Zhang, J.; Zen, K.; Zhang, C.-Y. Secreted MicroRNAs: A New Form of Intercellular Communication. Trends Cell Biol. 2012, 22, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L.; Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 5th ed.; W. H. Freeman: New York, NY, USA, 2002; ISBN 978-0-7167-3051-4. [Google Scholar]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond Aerobic Glycolysis: Transformed Cells Can Engage in Glutamine Metabolism That Exceeds the Requirement for Protein and Nucleotide Synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V. Rethinking the Warburg Effect with Myc Micromanaging Glutamine Metabolism. Cancer Res. 2010, 70, 859–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agbu, P.; Carthew, R.W. MicroRNA-Mediated Regulation of Glucose and Lipid Metabolism. Nat. Rev. Mol. Cell Biol. 2021, 22, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Jeet, V.; Clements, J.A.; Gunter, J.H.; Batra, J. Emergence of MicroRNAs as Key Players in Cancer Cell Metabolism. Clin. Chem. 2019, 65, 1090–1101. [Google Scholar] [CrossRef]
- Redis, R.S.; Calin, G.A. SnapShot: Non-Coding RNAs and Metabolism. Cell Metab. 2017, 25. [Google Scholar] [CrossRef]
- Rottiers, V.; Näär, A.M. MicroRNAs in Metabolism and Metabolic Disorders. Nat. Rev. Mol. Cell Biol. 2012, 13, 239–250. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Lum, J.J.; Bauer, D.E.; Kong, M.; Harris, M.H.; Li, C.; Lindsten, T.; Thompson, C.B. Growth Factor Regulation of Autophagy and Cell Survival in the Absence of Apoptosis. Cell 2005, 120, 237–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaglio, D.; Soldati, C.; Vanoni, M.; Alberghina, L.; Chiaradonna, F. Glutamine Deprivation Induces Abortive S-Phase Rescued by Deoxyribonucleotides in k-Ras Transformed Fibroblasts. PLoS ONE 2009, 4, e4715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc Regulates a Transcriptional Program That Stimulates Mitochondrial Glutaminolysis and Leads to Glutamine Addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecchio, E.; Caiazza, C.; Mimmi, S.; Avagliano, A.; Iaccino, E.; Brusco, T.; Nisticò, N.; Maisano, D.; Aloisio, A.; Quinto, I.; et al. Metabolites Profiling of Melanoma Interstitial Fluids Reveals Uridine Diphosphate as Potent Immune Modulator Capable of Limiting Tumor Growth. Front. Cell Dev. Biol. 2021, 9, 730726. [Google Scholar] [CrossRef] [PubMed]
- Strohecker, A.M.; Guo, J.Y.; Karsli-Uzunbas, G.; Price, S.M.; Chen, G.J.; Mathew, R.; McMahon, M.; White, E. Autophagy Sustains Mitochondrial Glutamine Metabolism and Growth of BrafV600E-Driven Lung Tumors. Cancer Discov. 2013, 3, 1272–1285. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy Suppresses Progression of K-Ras-Induced Lung Tumors to Oncocytomas and Maintains Lipid Homeostasis. Genes Dev. 2013, 27, 1447–1461. [Google Scholar] [CrossRef] [Green Version]
- Kaelin, W.G.; Ratcliffe, P.J. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Kim, J.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-Mediated Expression of Pyruvate Dehydrogenase Kinase: A Metabolic Switch Required for Cellular Adaptation to Hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Sun, X.-X.; Qian, D.Z.; Dai, M.-S. Molecular Crosstalk Between MYC and HIF in Cancer. Front. Cell Dev. Biol. 2020, 8, 590576. [Google Scholar] [CrossRef]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of Glucose Transporter 1 and Glycolytic Gene Expression by C-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. C-Myc Transactivation of LDH-A: Implications for Tumor Metabolism and Growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A Expression Uncovers a Link between Glycolysis, Mitochondrial Physiology, and Tumor Maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of Lactate Dehydrogenase a Induces Oxidative Stress and Inhibits Tumor Progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudreau, A.; Purkey, H.E.; Hitz, A.; Robarge, K.; Peterson, D.; Labadie, S.; Kwong, M.; Hong, R.; Gao, M.; Del Nagro, C.; et al. Metabolic Plasticity Underpins Innate and Acquired Resistance to LDHA Inhibition. Nat. Chem. Biol. 2016, 12, 779–786. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and Adaptive Immune Cells in the Tumor Microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leone, R.D.; Powell, J.D. Metabolism of Immune Cells in Cancer. Nat. Rev. Cancer 2020, 20, 516–531. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [Green Version]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional Polarization of Tumour-Associated Macrophages by Tumour-Derived Lactic Acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Scharping, N.E.; Rivadeneira, D.B.; Menk, A.V.; Vignali, P.D.A.; Ford, B.R.; Rittenhouse, N.L.; Peralta, R.; Wang, Y.; Wang, Y.; DePeaux, K.; et al. Mitochondrial Stress Induced by Continuous Stimulation under Hypoxia Rapidly Drives T Cell Exhaustion. Nat. Immunol. 2021, 22, 205–215. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA Therapeutics: Towards a New Era for the Management of Cancer and Other Diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.D.; Naldini, L. Exploiting and Antagonizing MicroRNA Regulation for Therapeutic and Experimental Applications. Nat. Rev. Genet. 2009, 10, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Segal, M.; Slack, F.J. Challenges Identifying Efficacious MiRNA Therapeutics for Cancer. Expert Opin. Drug Discov. 2020, 15, 987–992. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Kang, Y.-K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.-L.; Kim, T.-Y.; et al. Phase 1 Study of MRX34, a Liposomal MiR-34a Mimic, in Patients with Advanced Solid Tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef] [PubMed]
- Van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and Activity of MicroRNA-Loaded Minicells in Patients with Recurrent Malignant Pleural Mesothelioma: A First-in-Man, Phase 1, Open-Label, Dose-Escalation Study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Anastasiadou, E.; Seto, A.G.; Beatty, X.; Hermreck, M.; Gilles, M.-E.; Stroopinsky, D.; Pinter-Brown, L.C.; Pestano, L.; Marchese, C.; Avigan, D.; et al. Cobomarsen, an Oligonucleotide Inhibitor of MiR-155, Slows DLBCL Tumor Cell Growth In Vitro and In Vivo. Clin. Cancer Res. 2021, 27, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Teplyuk, N.M.; Uhlmann, E.J.; Gabriely, G.; Volfovsky, N.; Wang, Y.; Teng, J.; Karmali, P.; Marcusson, E.; Peter, M.; Mohan, A.; et al. Therapeutic Potential of Targeting MicroRNA-10b in Established Intracranial Glioblastoma: First Steps toward the Clinic. EMBO Mol. Med. 2016, 8, 268–287. [Google Scholar] [CrossRef]
- Telford, B.J.; Yahyanejad, S.; de Gunst, T.; den Boer, H.C.; Vos, R.M.; Stegink, M.; van den Bosch, M.T.J.; Alemdehy, M.F.; van Pinxteren, L.A.H.; Schaapveld, R.Q.J.; et al. Multi-Modal Effects of 1B3, a Novel Synthetic MiR-193a-3p Mimic, Support Strong Potential for Therapeutic Intervention in Oncology. Oncotarget 2021, 12, 422–439. [Google Scholar] [CrossRef]
- Van den Bosch, M.T.J.; Yahyanejad, S.; Alemdehy, M.F.; Telford, B.J.; de Gunst, T.; den Boer, H.C.; Vos, R.M.; Stegink, M.; van Pinxteren, L.A.H.; Schaapveld, R.Q.J.; et al. Transcriptome-Wide Analysis Reveals Insight into Tumor Suppressor Functions of 1B3, a Novel Synthetic MiR-193a-3p Mimic. Mol. Ther. Nucleic Acids 2021, 23, 1161–1171. [Google Scholar] [CrossRef]
- Hatziapostolou, M.; Polytarchou, C.; Iliopoulos, D. MiRNAs Link Metabolic Reprogramming to Oncogenesis. Trends Endocrinol. Metab. 2013, 24, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Zhang, L.-F.; Zhang, H.-W.; Hu, S.; Lu, M.-H.; Liang, S.; Li, B.; Li, Y.; Li, D.; Wang, E.-D.; et al. A Novel MiR-155/MiR-143 Cascade Controls Glycolysis by Regulating Hexokinase 2 in Breast Cancer Cells. EMBO J. 2012, 31, 1985–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Lee, E.; Jung, J.; Lee, J.W.; Kim, H.J.; Kim, J.; Yoo, H.J.; Lee, H.J.; Chae, S.Y.; Jeon, S.M.; et al. MicroRNA-155 Positively Regulates Glucose Metabolism via PIK3R1-FOXO3a-CMYC Axis in Breast Cancer. Oncogene 2018, 37, 2982–2991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, W.; Riopel, M.; Bandyopadhyay, G.; Dong, Y.; Birmingham, A.; Seo, J.B.; Ofrecio, J.M.; Wollam, J.; Hernandez-Carretero, A.; Fu, W.; et al. Adipose Tissue Macrophage-Derived Exosomal MiRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell 2017, 171, 372–384.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, H.; Liu, A.; Fang, C.; Hao, J.; Wang, Z. Lactate Dehydrogenase a Negatively Regulated by MiRNAs Promotes Aerobic Glycolysis and Is Increased in Colorectal Cancer. Oncotarget 2015, 6, 19456–19468. [Google Scholar] [CrossRef]
- Wu, J.; Bao, J.; Kim, M.; Yuan, S.; Tang, C.; Zheng, H.; Mastick, G.S.; Xu, C.; Yan, W. Two MiRNA Clusters, MiR-34b/c and MiR-449, Are Essential for Normal Brain Development, Motile Ciliogenesis, and Spermatogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, E2851–E2857. [Google Scholar] [CrossRef] [Green Version]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A MicroRNA Component of the P53 Tumour Suppressor Network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Liao, Y.; Tang, L. MicroRNA-34 Family: A Potential Tumor Suppressor and Therapeutic Candidate in Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 967–977. [Google Scholar] [CrossRef] [Green Version]
- Hermeking, H. The MiR-34 Family in Cancer and Apoptosis. Cell Death Differ. 2010, 17, 193–199. [Google Scholar] [CrossRef]
- Xiao, X.; Huang, X.; Ye, F.; Chen, B.; Song, C.; Wen, J.; Zhang, Z.; Zheng, G.; Tang, H.; Xie, X. The MiR-34a-LDHA Axis Regulates Glucose Metabolism and Tumor Growth in Breast Cancer. Sci. Rep. 2016, 6, 21735. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Xu, J.; Pan, X.; Zhang, Y.; Weng, Y.; Zhou, D.; He, S. LncRNA KCNQ1OT1 Sponges MiR-34c-5p to Promote Osteosarcoma Growth via ALDOA Enhanced Aerobic Glycolysis. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Simão, A.L.; Afonso, M.B.; Rodrigues, P.M.; Gama-Carvalho, M.; Machado, M.V.; Cortez-Pinto, H.; Rodrigues, C.M.P.; Castro, R.E. Skeletal Muscle MiR-34a/SIRT1:AMPK Axis Is Activated in Experimental and Human Non-Alcoholic Steatohepatitis. J. Mol. Med. 2019, 97, 1113–1126. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Cao, X.; Zhang, W.; Pan, G.; Yi, Q.; Zhong, W.; Yan, D. MicroRNA-31-5p Enhances the Warburg Effect via Targeting FIH. FASEB J. 2019, 33, 545–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charostad, J.; Nakhaie, M.; Dehghani, A.; Faghihloo, E. The Interplay between EBV and KSHV Viral Products and NF-ΚB Pathway in Oncogenesis. Infect. Agents Cancer 2020, 15, 62. [Google Scholar] [CrossRef] [PubMed]
- Yogev, O.; Lagos, D.; Enver, T.; Boshoff, C. Kaposi’s Sarcoma Herpesvirus MicroRNAs Induce Metabolic Transformation of Infected Cells. PLoS Pathog. 2014, 10, e1004400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Zhou, Y.; Zhang, L.; Chen, Y.; Lyu, X.; Cai, L.; Lu, Y.; Deng, Y.; Wang, J.; Yao, K.; et al. EBV-MiR-BART1 is Involved in Regulating Metabolism-Associated Genes in Nasopharyngeal Carcinoma. Biochem. Biophys. Res. Commun. 2013, 436, 19–24. [Google Scholar] [CrossRef]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate Dehydrogenase Diverts Glycolytic Flux and Contributes to Oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Wiggins, J.F.; Ruffino, L.; Kelnar, K.; Omotola, M.; Patrawala, L.; Brown, D.; Bader, A.G. Development of a Lung Cancer Therapeutic Based on the Tumor Suppressor MicroRNA-34. Cancer Res. 2010, 70, 5923–5930. [Google Scholar] [CrossRef] [Green Version]
- Reid, G.; Pel, M.E.; Kirschner, M.B.; Cheng, Y.Y.; Mugridge, N.; Weiss, J.; Williams, M.; Wright, C.; Edelman, J.J.B.; Vallely, M.P.; et al. Restoring Expression of MiR-16: A Novel Approach to Therapy for Malignant Pleural Mesothelioma. Ann. Oncol. 2013, 24, 3128–3135. [Google Scholar] [CrossRef]
- Huang, X.; Hou, Y.; Weng, X.; Pang, W.; Hou, L.; Liang, Y.; Wang, Y.; Du, L.; Wu, T.; Yao, M.; et al. Diethyldithiocarbamate-Copper Complex (CuET) Inhibits Colorectal Cancer Progression via MiR-16-5p and 15b-5p/ALDH1A3/PKM2 Axis-Mediated Aerobic Glycolysis Pathway. Oncogenesis 2021, 10, 1–16. [Google Scholar] [CrossRef]
- Ye, T.; Liang, Y.; Zhang, D.; Zhang, X. MicroRNA-16-1-3p Represses Breast Tumor Growth and Metastasis by Inhibiting PGK1-Mediated Warburg Effect. Front. Cell Dev. Biol. 2020, 8, 615154. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Wang, Y.; Shi, Z.; Liu, J.; Sun, P.; Hou, X.; Zhang, J.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic Reprogramming of Cancer-Associated Fibroblasts by IDH3α Downregulation. Cell Rep. 2015, 10, 1335–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilinc, S.; Paisner, R.; Camarda, R.; Gupta, S.; Momcilovic, O.; Kohnz, R.A.; Avsaroglu, B.; L’Etoile, N.D.; Perera, R.M.; Nomura, D.K.; et al. Oncogene-Regulated Release of Extracellular Vesicles. Dev. Cell 2021, 56, 1989–2006.e6. [Google Scholar] [CrossRef]
- Li, X.; Wenes, M.; Romero, P.; Huang, S.C.-C.; Fendt, S.-M.; Ho, P.-C. Navigating Metabolic Pathways to Enhance Antitumour Immunity and Immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 425–441. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin Inhibits Mitochondrial Complex I of Cancer Cells to Reduce Tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Pedroza-Torres, A.; Romero-Córdoba, S.L.; Justo-Garrido, M.; Salido-Guadarrama, I.; Rodríguez-Bautista, R.; Montaño, S.; Muñiz-Mendoza, R.; Arriaga-Canon, C.; Fragoso-Ontiveros, V.; Álvarez-Gómez, R.M.; et al. MicroRNAs in Tumor Cell Metabolism: Roles and Therapeutic Opportunities. Front. Oncol. 2019, 9, 1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, E.; Maj, T.; Kryczek, I.; Li, W.; Wu, K.; Zhao, L.; Wei, S.; Crespo, J.; Wan, S.; Vatan, L.; et al. Cancer Mediates Effector T Cell Dysfunction by Targeting MicroRNAs and EZH2 via Glycolysis Restriction. Nat. Immunol. 2016, 17, 95–103. [Google Scholar] [CrossRef]
- Mathis, D.; Shoelson, S.E. Immunometabolism: An Emerging Frontier. Nat. Rev. Immunol. 2011, 11, 81–83. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A Guide to Immunometabolism for Immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Almeida, L.; Lochner, M.; Berod, L.; Sparwasser, T. Metabolic Pathways in T Cell Activation and Lineage Differentiation. Semin. Immunol. 2016, 28, 514–524. [Google Scholar] [CrossRef] [Green Version]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The CD28 Signaling Pathway Regulates Glucose Metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, S.R.; Herman, C.E.; Maciver, N.J.; Wofford, J.A.; Wieman, H.L.; Hammen, J.J.; Rathmell, J.C. Glucose Uptake Is Limiting in T Cell Activation and Requires CD28-Mediated Akt-Dependent and Independent Pathways. J. Immunol. 2008, 180, 4476–4486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroesen, B.-J.; Teteloshvili, N.; Smigielska-Czepiel, K.; Brouwer, E.; Boots, A.M.H.; van den Berg, A.; Kluiver, J. Immuno-MiRs: Critical Regulators of T-Cell Development, Function and Ageing. Immunology 2015, 144, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Song, Z.; Wang, B.; Zhang, J.-A. Emerging Roles of MicroRNAs in the Metabolic Control of Immune Cells. Cancer Lett. 2018, 433, 10–17. [Google Scholar] [CrossRef]
- King, B.C.; Esguerra, J.L.S.; Golec, E.; Eliasson, L.; Kemper, C.; Blom, A.M. CD46 Activation Regulates MiR-150-Mediated Control of GLUT1 Expression and Cytokine Secretion in Human CD4+ T Cells. J. Immunol. 2016, 196, 1636–1645. [Google Scholar] [CrossRef] [Green Version]
- Fang, R.; Xiao, T.; Fang, Z.; Sun, Y.; Li, F.; Gao, Y.; Feng, Y.; Li, L.; Wang, Y.; Liu, X.; et al. MicroRNA-143 (MiR-143) Regulates Cancer Glycolysis via Targeting Hexokinase 2 Gene*. J. Biol. Chem. 2012, 287, 23227–23235. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhang, Z.; Li, F.; Ping, Y.; Qin, G.; Zhang, C.; Zhang, Y. MiR-143 Regulates Memory T Cell Differentiation by Reprogramming T Cell Metabolism. J. Immunol. 2018, 201, 2165–2175. [Google Scholar] [CrossRef]
- Barré-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vézinet-Brun, F.; Rouzioux, C.; et al. Isolation of a T-Lymphotropic Retrovirus from a Patient at Risk for Acquired Immune Deficiency Syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar] [CrossRef] [Green Version]
- Gallo, R.C.; Sarin, P.S.; Gelmann, E.P.; Robert-Guroff, M.; Richardson, E.; Kalyanaraman, V.S.; Mann, D.; Sidhu, G.D.; Stahl, R.E.; Zolla-Pazner, S.; et al. Isolation of Human T-Cell Leukemia Virus in Acquired Immune Deficiency Syndrome (AIDS). Science 1983, 220, 865–867. [Google Scholar] [CrossRef]
- Deeks, S.G.; Overbaugh, J.; Phillips, A.; Buchbinder, S. HIV Infection. Nat. Rev. Dis. Primers 2015, 1, 15035. [Google Scholar] [CrossRef]
- Clavel, F.; Guétard, D.; Brun-Vézinet, F.; Chamaret, S.; Rey, M.A.; Santos-Ferreira, M.O.; Laurent, A.G.; Dauguet, C.; Katlama, C.; Rouzioux, C. Isolation of a New Human Retrovirus from West African Patients with AIDS. Science 1986, 233, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Marlink, R.; Kanki, P.; Thior, I.; Travers, K.; Eisen, G.; Siby, T.; Traore, I.; Hsieh, C.C.; Dia, M.C.; Gueye, E.H.; et al. Reduced Rate of Disease Development after HIV-2 Infection as Compared to HIV-1. Science 1994, 265, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Dalgleish, A.G.; Beverley, P.C.L.; Clapham, P.R.; Crawford, D.H.; Greaves, M.F.; Weiss, R.A. The CD4 (T4) Antigen Is an Essential Component of the Receptor for the AIDS Retrovirus. Nature 1984, 312, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, M.; Stanwick, T.L.; Dempsey, M.P.; Lamonica, C.A. HIV-1 Replication Is Controlled at the Level of T Cell Activation and Proviral Integration. EMBO J. 1990, 9, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Buzon, M.J.; Sun, H.; Li, C.; Shaw, A.; Seiss, K.; Ouyang, Z.; Martin-Gayo, E.; Leng, J.; Henrich, T.J.; Li, J.Z.; et al. HIV-1 Persistence in CD4+ T Cells with Stem Cell-like Properties. Nat. Med. 2014, 20, 139–142. [Google Scholar] [CrossRef]
- Goodwin, C.M.; Xu, S.; Munger, J. Stealing the Keys to the Kitchen: Viral Manipulation of the Host Cell Metabolic Network. Trends Microbiol. 2015, 23, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Valle-Casuso, J.C.; Angin, M.; Volant, S.; Passaes, C.; Monceaux, V.; Mikhailova, A.; Bourdic, K.; Avettand-Fenoel, V.; Boufassa, F.; Sitbon, M.; et al. Cellular Metabolism Is a Major Determinant of HIV-1 Reservoir Seeding in CD4+ T Cells and Offers an Opportunity to Tackle Infection. Cell Metab. 2019, 29, 611–626.e5. [Google Scholar] [CrossRef] [Green Version]
- Clerc, I.; Abba Moussa, D.; Vahlas, Z.; Tardito, S.; Oburoglu, L.; Hope, T.J.; Sitbon, M.; Dardalhon, V.; Mongellaz, C.; Taylor, N. Entry of Glucose- and Glutamine-Derived Carbons into the Citric Acid Cycle Supports Early Steps of HIV-1 Infection in CD4 T Cells. Nat. Metab. 2019, 1, 717–730. [Google Scholar] [CrossRef]
- Zack, J.A.; Kim, S.G.; Vatakis, D.N. HIV Restriction in Quiescent CD4+T Cells. Retrovirology 2013, 10, 37. [Google Scholar] [CrossRef] [Green Version]
- Loisel-Meyer, S.; Swainson, L.; Craveiro, M.; Oburoglu, L.; Mongellaz, C.; Costa, C.; Martinez, M.; Cosset, F.-L.; Battini, J.-L.; Herzenberg, L.A.; et al. Glut1-Mediated Glucose Transport Regulates HIV Infection. Proc. Natl. Acad. Sci. USA 2012, 109, 2549–2554. [Google Scholar] [CrossRef] [Green Version]
- Taylor, H.E.; Calantone, N.; Lichon, D.; Hudson, H.; Clerc, I.; Campbell, E.M.; D’Aquila, R.T. MTOR Overcomes Multiple Metabolic Restrictions to Enable HIV-1 Reverse Transcription and Intracellular Transport. Cell Rep. 2020, 31, 107810. [Google Scholar] [CrossRef] [PubMed]
- Heredia, A.; Le, N.; Gartenhaus, R.B.; Sausville, E.; Medina-Moreno, S.; Zapata, J.C.; Davis, C.; Gallo, R.C.; Redfield, R.R. Targeting of MTOR Catalytic Site Inhibits Multiple Steps of the HIV-1 Lifecycle and Suppresses HIV-1 Viremia in Humanized Mice. Proc. Natl. Acad. Sci. USA 2015, 112, 9412–9417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishimoto, N.; Yamamoto, K.; Abe, T.; Yasuoka, N.; Takamune, N.; Misumi, S. Glucose-Dependent Aerobic Glycolysis Contributes to Recruiting Viral Components into HIV-1 Particles to Maintain Infectivity. Biochem. Biophys. Res. Commun. 2021, 549, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, A.; Kavanagh Williamson, M.; Huthoff, H. HIV-1 Pathogenicity and Virion Production Are Dependent on the Metabolic Phenotype of Activated CD4+ T Cells. Retrovirology 2014, 11, 98. [Google Scholar] [CrossRef] [Green Version]
- Amie, S.M.; Noble, E.; Kim, B. Intracellular Nucleotide Levels and the Control of Retroviral Infections. Virology 2013, 436, 247–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. SAMHD1 Restricts the Replication of Human Immunodeficiency Virus Type 1 by Depleting the Intracellular Pool of Deoxynucleoside Triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, M.; Ratner, L. Myristoylation-Dependent Replication and Assembly of Human Immunodeficiency Virus 1. Proc. Natl. Acad. Sci. USA 1990, 87, 523–527. [Google Scholar] [CrossRef] [Green Version]
- Ono, A.; Freed, E.O. Plasma Membrane Rafts Play a Critical Role in HIV-1 Assembly and Release. Proc. Natl. Acad. Sci. USA 2001, 98, 13925–13930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Lint, C.; Emiliani, S.; Ott, M.; Verdin, E. Transcriptional Activation and Chromatin Remodeling of the HIV-1 Promoter in Response to Histone Acetylation. EMBO J. 1996, 15, 1112–1120. [Google Scholar] [CrossRef]
- Kang, S.; Tang, H. HIV-1 Infection and Glucose Metabolism Reprogramming of T Cells: Another Approach Toward Functional Cure and Reservoir Eradication. Front. Immunol. 2020, 11, 2621. [Google Scholar] [CrossRef]
- Sáez-Cirión, A.; Sereti, I. Immunometabolism and HIV-1 Pathogenesis: Food for Thought. Nat. Rev. Immunol. 2021, 21, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh Williamson, M.; Coombes, N.; Juszczak, F.; Athanasopoulos, M.; Khan, M.B.; Eykyn, T.R.; Srenathan, U.; Taams, L.S.; Dias Zeidler, J.; Da Poian, A.T.; et al. Upregulation of Glucose Uptake and Hexokinase Activity of Primary Human CD4+ T Cells in Response to Infection with HIV-1. Viruses 2018, 10, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, C.S.; Ostrowski, M.; Gouillou, M.; Tsai, L.; Yu, D.; Zhou, J.; Henstridge, D.C.; Maisa, A.; Hearps, A.C.; Lewin, S.R.; et al. Increased Glucose Metabolic Activity Is Associated with CD4+ T-Cell Activation and Depletion during Chronic HIV Infection. AIDS 2014, 28, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wang, Q.; Ghneim, K.; Wang, L.; Rampanelli, E.; Holley-Guthrie, E.; Cheng, L.; Garrido, C.; Margolis, D.M.; Eller, L.A.; et al. Multi-Omics Analyses Reveal That HIV-1 Alters CD4 + T Cell Immunometabolism to Fuel Virus Replication. Nat. Immunol. 2021, 22, 423–433. [Google Scholar] [CrossRef]
- Shytaj, I.L.; Procopio, F.A.; Tarek, M.; Carlon-Andres, I.; Tang, H.-Y.; Goldman, A.R.; Munshi, M.; Pal, V.K.; Forcato, M.; Sreeram, S.; et al. Glycolysis Downregulation is a Hallmark of HIV-1 Latency and Sensitizes Infected Cells to Oxidative Stress. EMBO Mol. Med. 2021, 13, e13901. [Google Scholar] [CrossRef]
- Balasubramaniam, M.; Pandhare, J.; Dash, C. Are MicroRNAs Important Players in HIV-1 Infection? An Update. Viruses 2018, 10, 110. [Google Scholar] [CrossRef] [Green Version]
- Amaral, A.J.; Andrade, J.; Foxall, R.B.; Matoso, P.; Matos, A.M.; Soares, R.S.; Rocha, C.; Ramos, C.G.; Tendeiro, R.; Serra-Caetano, A.; et al. MiRNA Profiling of Human Naive CD4 T Cells Links MiR-34c-5p to Cell Activation and HIV Replication. EMBO J. 2017, 36, 346–360. [Google Scholar] [CrossRef] [Green Version]
- Ruelas, D.S.; Chan, J.K.; Oh, E.; Heidersbach, A.J.; Hebbeler, A.M.; Chavez, L.; Verdin, E.; Rape, M.; Greene, W.C. MicroRNA-155 Reinforces HIV Latency. J. Biol. Chem. 2015, 290, 13736–13748. [Google Scholar] [CrossRef] [Green Version]
- Heinson, A.I.; Woo, J.; Mukim, A.; White, C.H.; Moesker, B.; Bosque, A.; Spina, C.A.; Woelk, C.H.; Macarthur, B.D.; Beliakova-Bethell, N. Micro RNA Targets in HIV Latency: Insights into Novel Layers of Latency Control. AIDS Res. Hum. Retrovir. 2021, 37, 109–121. [Google Scholar] [CrossRef]
- Huang, J.; Wang, F.; Argyris, E.; Chen, K.; Liang, Z.; Tian, H.; Huang, W.; Squires, K.; Verlinghieri, G.; Zhang, H. Cellular MicroRNAs Contribute to HIV-1 Latency in Resting Primary CD4+ T Lymphocytes. Nat. Med. 2007, 13, 1241–1247. [Google Scholar] [CrossRef]
- Jin, C.; Peng, X.; Liu, F.; Cheng, L.; Lu, X.; Yao, H.; Wu, H.; Wu, N. MicroRNA-181 Expression Regulates Specific Post-Transcriptional Level of SAMHD1 Expression in Vitro. Biochem. Biophys. Res. Commun. 2014, 452, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Triboulet, R.; Mari, B.; Lin, Y.-L.; Chable-Bessia, C.; Bennasser, Y.; Lebrigand, K.; Cardinaud, B.; Maurin, T.; Barbry, P.; Baillat, V.; et al. Suppression of MicroRNA-Silencing Pathway by HIV-1 during Virus Replication. Science 2007, 315, 1579–1582. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.K.; Sengupta, P.; Waki, K.; Engelenburg, S.B.V.; Ochiya, T.; Ablan, S.D.; Freed, E.O.; Lippincott-Schwartz, J. MicroRNA Binding to the HIV-1 Gag Protein Inhibits Gag Assembly and Virus Production. Proc. Natl. Acad. Sci. USA 2014, 111, E2676–E2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathans, R.; Chu, C.-Y.; Serquina, A.K.; Lu, C.-C.; Cao, H.; Rana, T.M. Cellular MicroRNA and P Bodies Modulate Host-HIV-1 Interactions. Mol. Cell 2009, 34, 696–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, K.; Sung, T.-L.; Rice, A.P. Regulation of Cyclin T1 and HIV-1 Replication by MicroRNAs in Resting CD4+ T Lymphocytes. J. Virol. 2012, 86, 3244–3252. [Google Scholar] [CrossRef] [Green Version]
- Dubey, R.C.; Alam, N.B.; Gaur, R. MiR-150-Mediated Increase in Glucose Uptake in HIV-Infected Cells. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of Hepatitis C Virus RNA Abundance by a Liver-Specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [Green Version]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. MiR-122 Regulation of Lipid Metabolism Revealed by in Vivo Antisense Targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Norman, K.L.; Sarnow, P. Modulation of Hepatitis C Virus RNA Abundance and the Isoprenoid Biosynthesis Pathway by MicroRNA MiR-122 Involves Distinct Mechanisms. J. Virol. 2010, 84, 666–670. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.M.; Xu, Z.; Shek, F.H.; Wong, K.-F.; Lee, N.P.; Poon, R.T.; Chen, J.; Luk, J.M. MiR-122 Targets Pyruvate Kinase M2 and Affects Metabolism of Hepatocellular Carcinoma. PLoS ONE 2014, 9, e86872. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, D.; Cassel, T.; Teng, K.-Y.; Aljuhani, M.; Chowdhary, V.K.; Hu, P.; Zhang, X.; Fan, T.W.-M.; Ghoshal, K. Regulation of Hepatic Glutamine Metabolism by MiR-122. Mol. Metab. 2020, 34, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Shirasaki, T.; Honda, M.; Shimakami, T.; Horii, R.; Yamashita, T.; Sakai, Y.; Sakai, A.; Okada, H.; Watanabe, R.; Murakami, S.; et al. MicroRNA-27a Regulates Lipid Metabolism and Inhibits Hepatitis C Virus Replication in Human Hepatoma Cells. J. Virol. 2013, 87, 5270–5286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, T.A.; Singaravelu, R.; Powdrill, M.H.; Nhan, J.; Ahmed, N.; Özcelik, D.; Pezacki, J.P. MicroRNA-124 Regulates Fatty Acid and Triglyceride Homeostasis. iScience 2018, 10, 149–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCaskill, J.L.; Ressel, S.; Alber, A.; Redford, J.; Power, U.F.; Schwarze, J.; Dutia, B.M.; Buck, A.H. Broad-Spectrum Inhibition of Respiratory Virus Infection by MicroRNA Mimics Targeting P38 MAPK Signaling. Mol. Ther. Nucleic Acids 2017, 7, 256–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farberov, L.; Herzig, E.; Modai, S.; Isakov, O.; Hizi, A.; Shomron, N. MicroRNA-Mediated Regulation of P21 and TASK1 Cellular Restriction Factors Enhances HIV-1 Infection. J. Cell Sci. 2015, 128, 1607–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, C.-J.; Jia, Y.-H.; Tian, R.-R.; Ding, M.; Zhang, C.; Wang, J.-H. Translation of Pur-α Is Targeted by Cellular MiRNAs to Modulate the Differentiation-Dependent Susceptibility of Monocytes to HIV-1 Infection. FASEB J. 2012, 26, 4755–4764. [Google Scholar] [CrossRef] [Green Version]
- Kukoyi, A.T.; Fan, X.; Staitieh, B.S.; Hybertson, B.M.; Gao, B.; McCord, J.M.; Guidot, D.M. MiR-144 Mediates Nrf2 Inhibition and Alveolar Epithelial Dysfunction in HIV-1 Transgenic Rats. Am. J. Physiol. Cell Physiol. 2019, 317, C390–C397. [Google Scholar] [CrossRef]
- Swaminathan, G.; Rossi, F.; Sierra, L.-J.; Gupta, A.; Navas-Martín, S.; Martín-García, J. A Role for MicroRNA-155 Modulation in the Anti-HIV-1 Effects of Toll-Like Receptor 3 Stimulation in Macrophages. PLoS Pathog. 2012, 8, e1002937. [Google Scholar] [CrossRef] [Green Version]
- Houzet, L.; Klase, Z.; Yeung, M.L.; Wu, A.; Le, S.-Y.; Quiñones, M.; Jeang, K.-T. The Extent of Sequence Complementarity Correlates with the Potency of Cellular MiRNA-Mediated Restriction of HIV-1. Nucleic Acids Res. 2012, 40, 11684–11696. [Google Scholar] [CrossRef] [Green Version]
- Jin, C.; Peng, X.; Liu, F.; Cheng, L.; Xie, T.; Lu, X.; Wu, H.; Wu, N. Interferon-Induced Sterile α Motif and Histidine/Aspartic Acid Domain-Containing Protein 1 Expression in Astrocytes and Microglia Is Mediated by MicroRNA-181a. AIDS 2016, 30, 2053–2064. [Google Scholar] [CrossRef]
- Pilakka-Kanthikeel, S.; Raymond, A.; Atluri, V.S.R.; Sagar, V.; Saxena, S.K.; Diaz, P.; Chevelon, S.; Concepcion, M.; Nair, M. Sterile α Motif and Histidine/Aspartic Acid Domain-Containing Protein 1 (SAMHD1)-Facilitated HIV Restriction in Astrocytes Is Regulated by MiRNA-181a. J. Neuroinflamm. 2015, 12, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bantug, G.R.; Galluzzi, L.; Kroemer, G.; Hess, C. The Spectrum of T Cell Metabolism in Health and Disease. Nat. Rev. Immunol. 2018, 18, 19–34. [Google Scholar] [CrossRef] [PubMed]
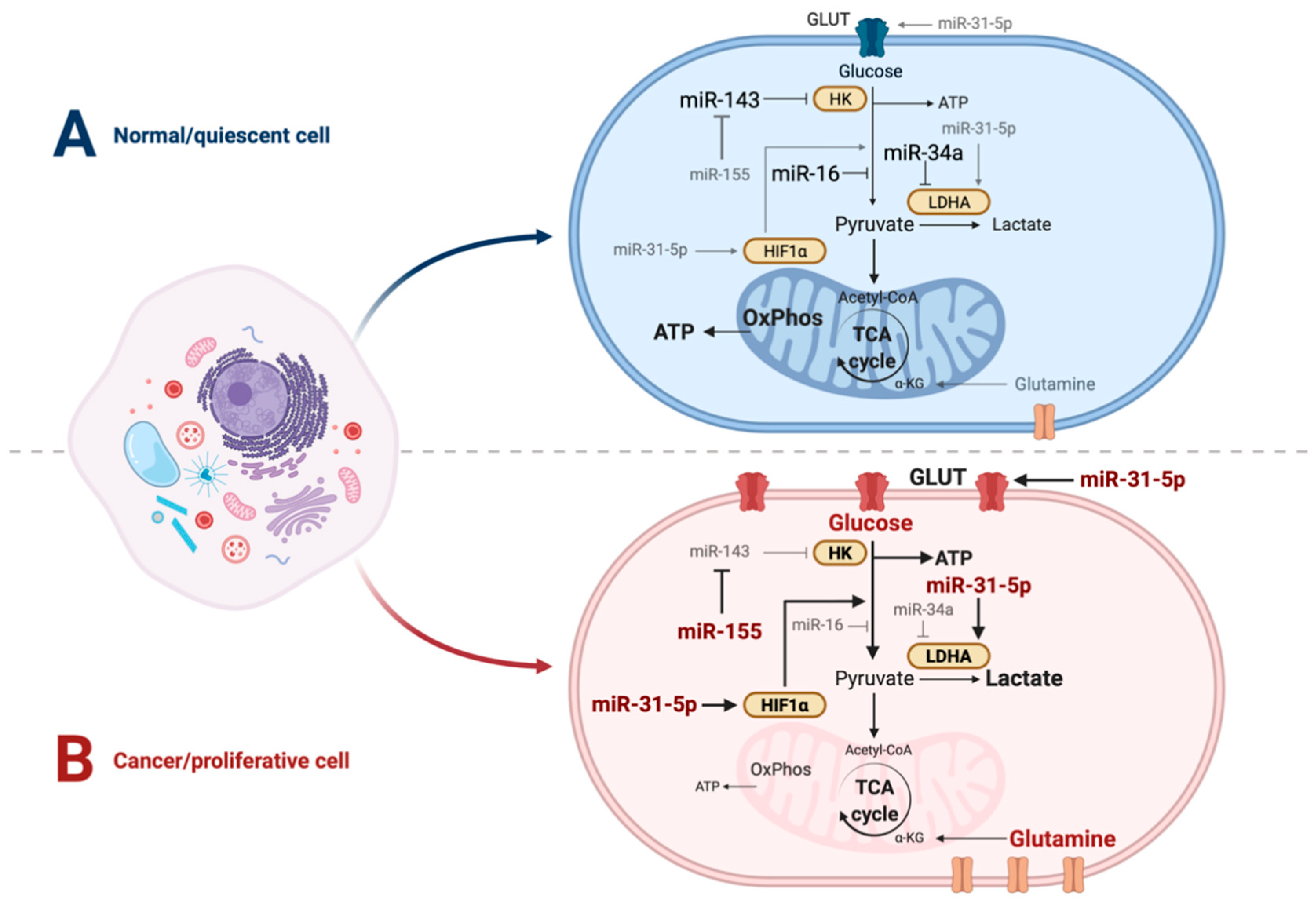

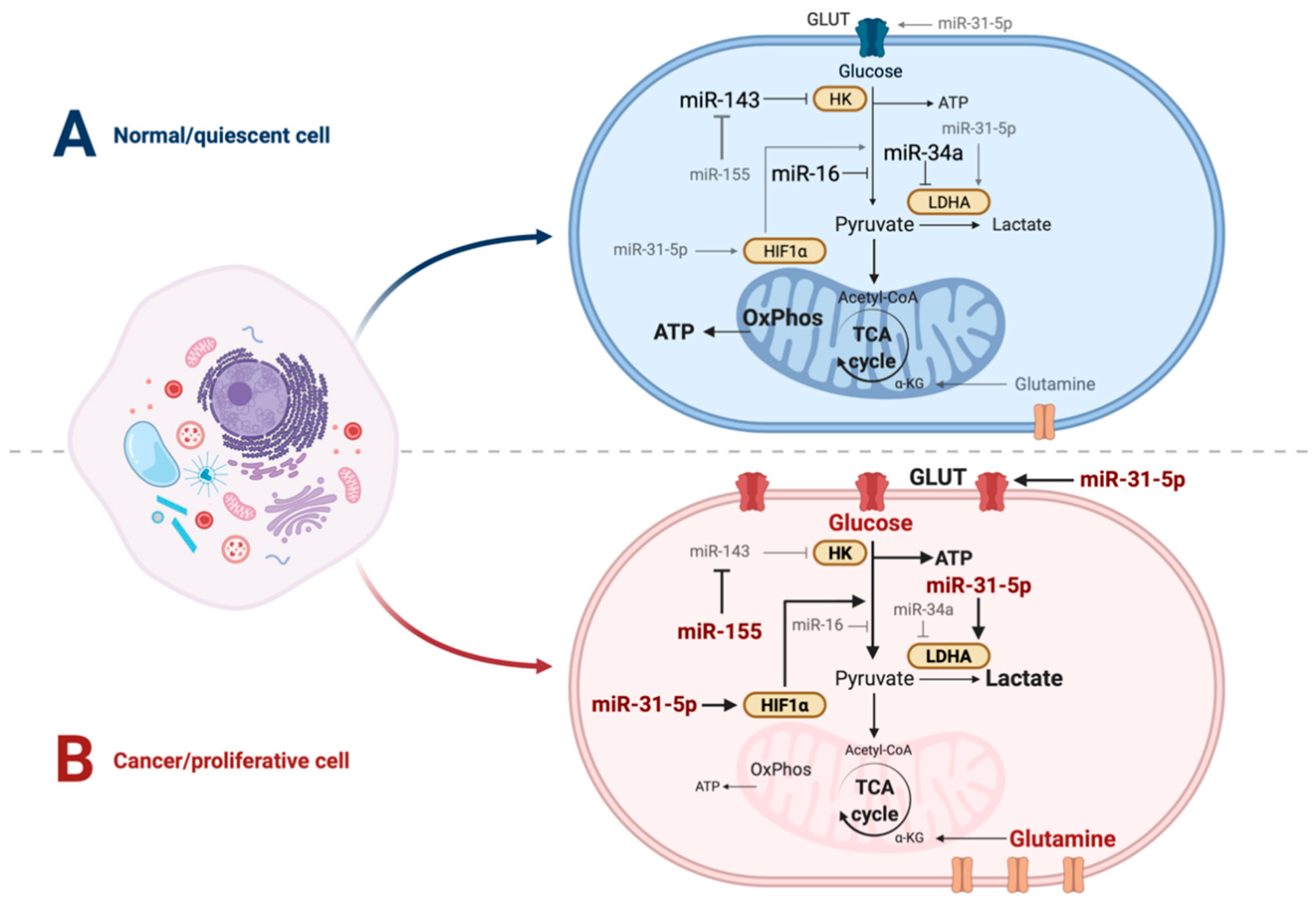{kind=link}
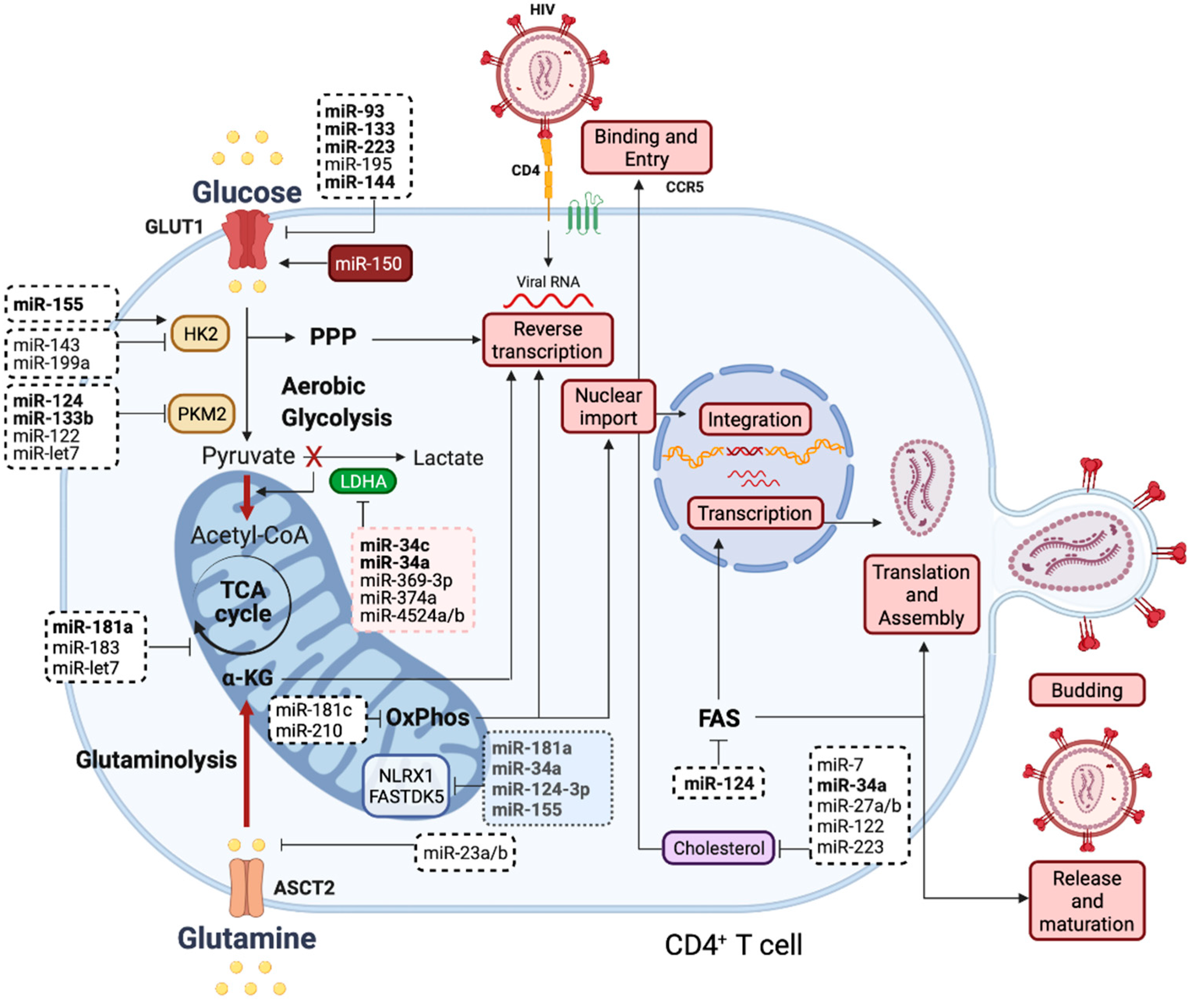
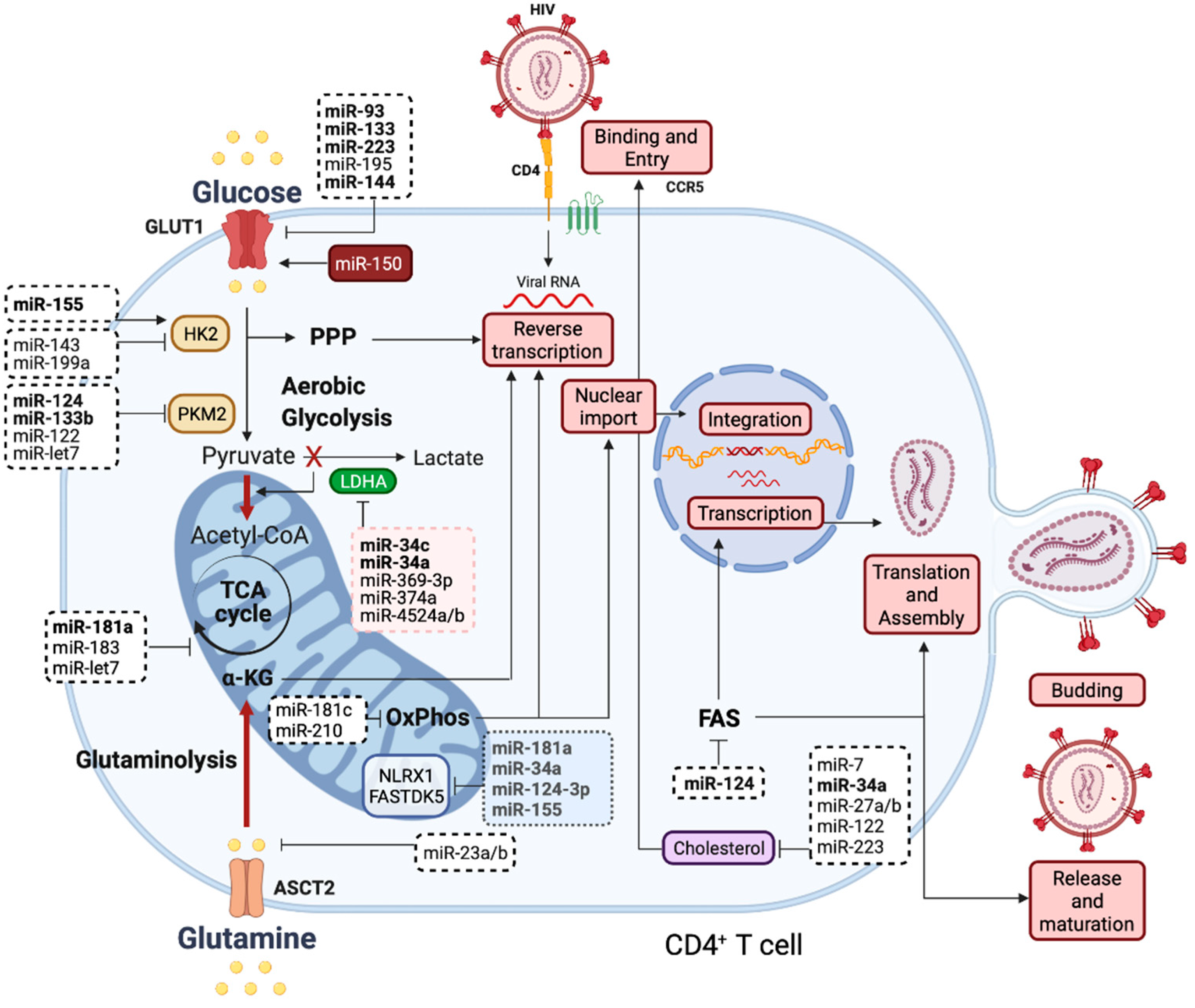{kind=link}
| Name | Company | miRNA (form) | Target Disease | Target mRNAs Regulating Metabolism | Clinical Trial I.D. | Reference |
|---|---|---|---|---|---|---|
| MRX34 | Synlogic | miR-34a (mimic) | Various solid tumors | LDHA (direct) | NCT01829971 | [45] |
| MesomiR−1 | EnGeneIC | miR-16 (mimic) | Malignant mesothelioma | PKM2 via ALDH1A3; PGK1 (direct) | NCT02369198 | [46] |
| Cobomarsen | Viridian | miR-155 (antagomiR) | Diffuse Large B-cell Lymphoma | HK2 via miR-143 | NCT02580552 | [47] |
| RGLS5579 | Regulus | miR-10b (antagomiR) | Glioblastoma | None reported | preclinical | [48] |
| INT 1B3 | InteRNA Technologies | miR-193a-3p (mimic) | Solid tumors | None reported | NCT04675996 | [49,50] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gibson, M.S.; Noronha-Estima, C.; Gama-Carvalho, M. Therapeutic Metabolic Reprograming Using microRNAs: From Cancer to HIV Infection. Genes 2022, 13, 273. https://doi.org/10.3390/genes13020273
Gibson MS, Noronha-Estima C, Gama-Carvalho M. Therapeutic Metabolic Reprograming Using microRNAs: From Cancer to HIV Infection. Genes. 2022; 13(2):273. https://doi.org/10.3390/genes13020273
Chicago/Turabian StyleGibson, Mark S., Cláudia Noronha-Estima, and Margarida Gama-Carvalho. 2022. "Therapeutic Metabolic Reprograming Using microRNAs: From Cancer to HIV Infection" Genes 13, no. 2: 273. https://doi.org/10.3390/genes13020273
APA StyleGibson, M. S., Noronha-Estima, C., & Gama-Carvalho, M. (2022). Therapeutic Metabolic Reprograming Using microRNAs: From Cancer to HIV Infection. Genes, 13(2), 273. https://doi.org/10.3390/genes13020273