
Snapshot of Mycobacterium tuberculosis Phylogenetics from an Indian State of Arunachal Pradesh Bordering China

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Setting and Sample Collection
2.2. Decontamination of Samples and Bactec MGIT 960 Culture Inoculation
2.3. Identification of Cultures Using In-House Multiplex PCR
2.4. Bactec MGIT 960 SIRE DST
2.5. Second Line DST Using Bactec MGIT 960
2.6. Whole Genome Sequencing
2.7. Identification of SNPs
2.8. Assignment of Principal Genetic Groups
2.9. Identification of Lineages and Sub-Lineages Using WGS SNP Barcoding
2.10. Phylogenetic Analysis and Construction of cgMLST
2.11. Data Analysis
3. Results
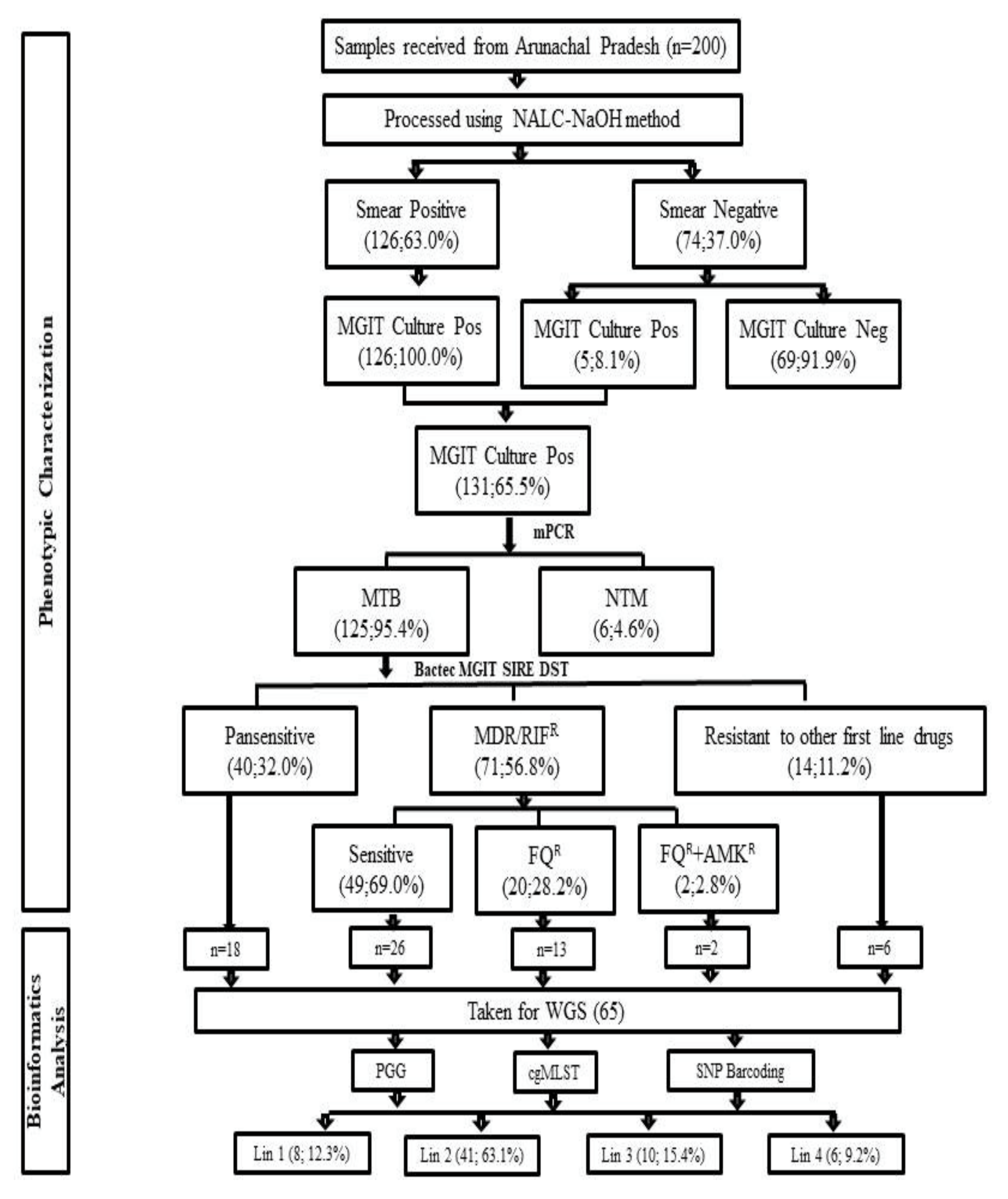
3.1. Demographic Details and Characteristics of MDR-TB Patients
3.2. Bactec MGIT 960 Culture Results and Identification M. tuberculosis Complex Isolates
3.3. Bactec MGIT 960 SIRE DST
3.4. Bactec MGIT 960 Second Line DST
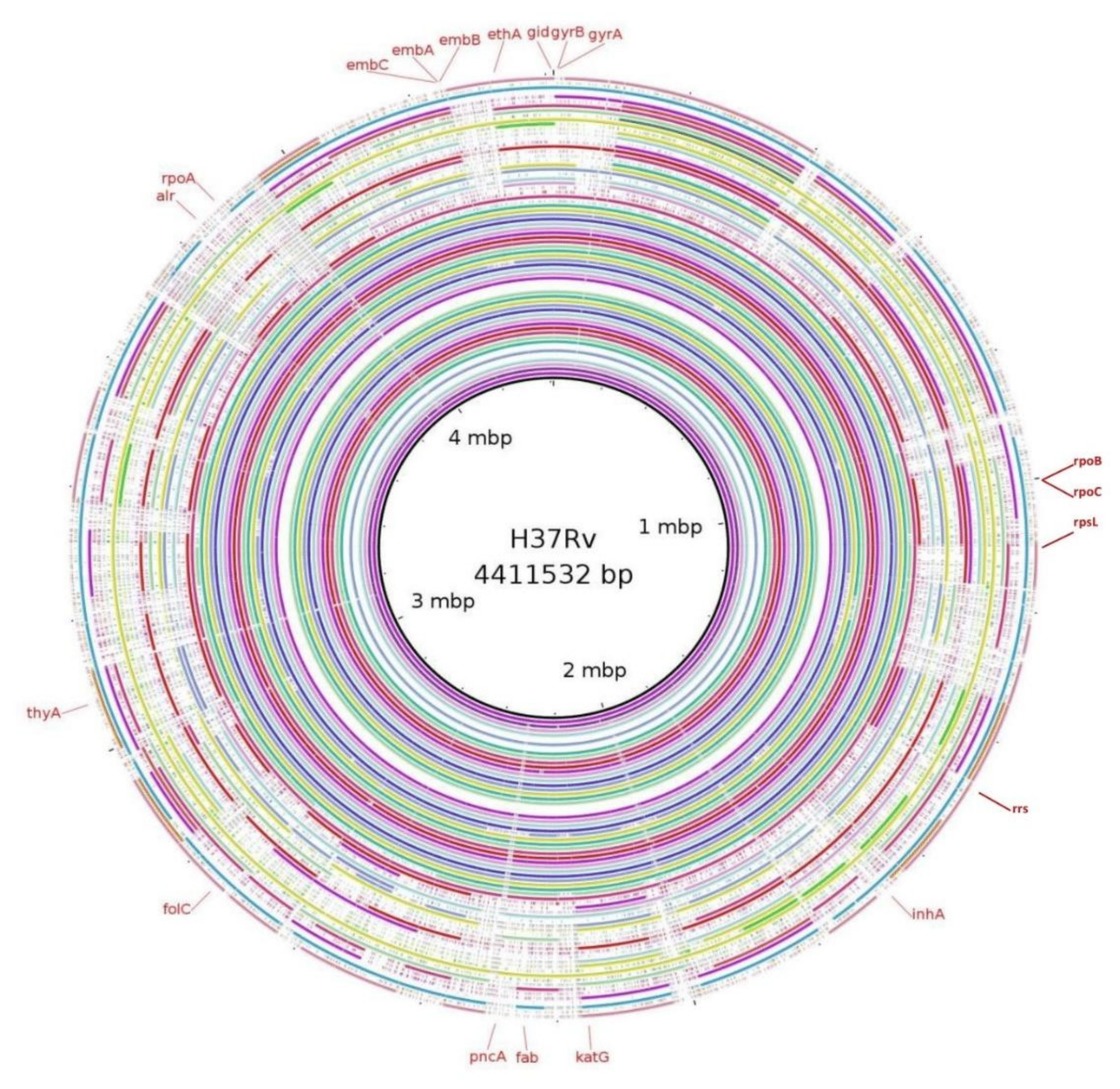
3.5. Mutations in Genes Associated with First- and Second-Line Drugs Using WGS
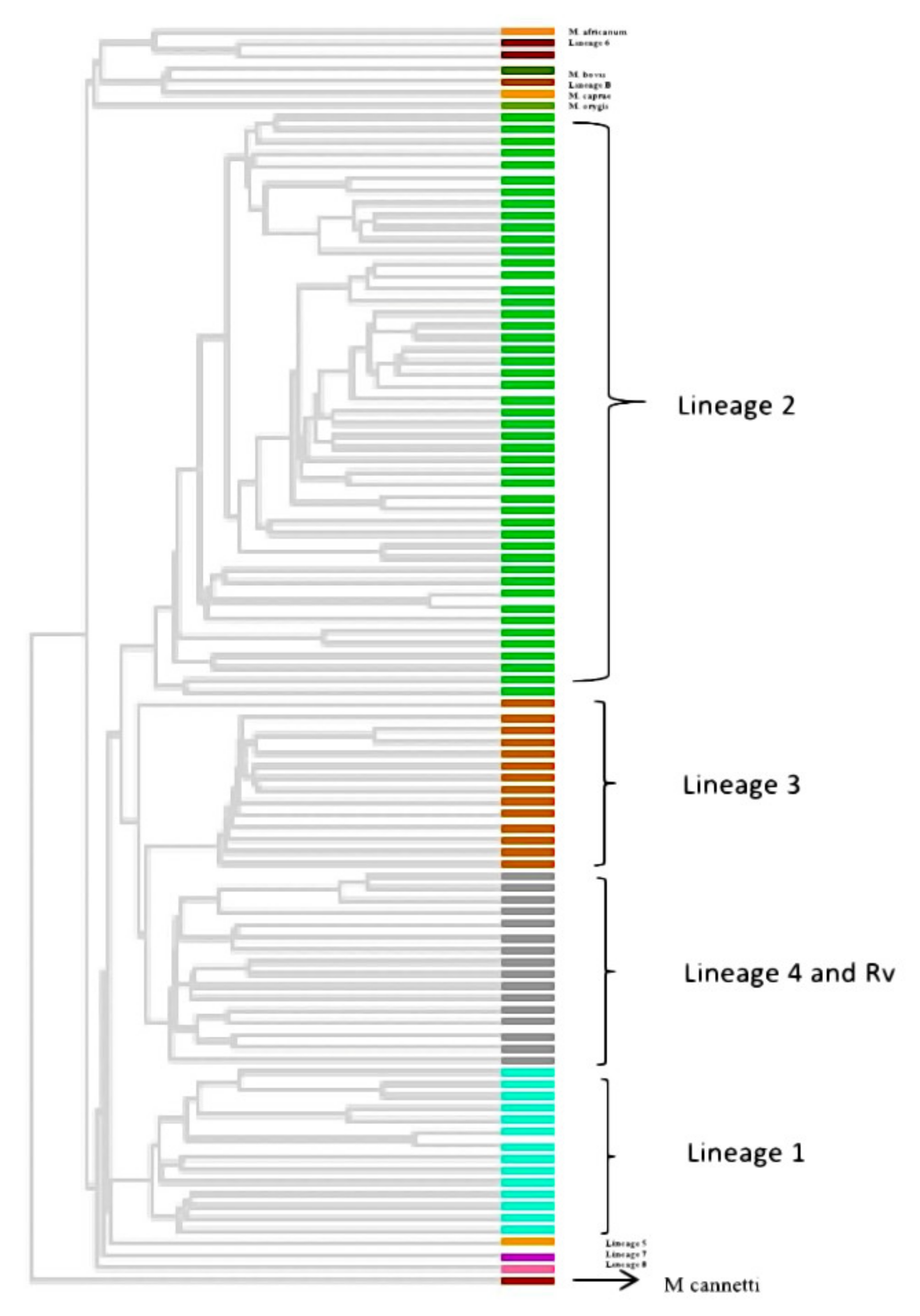
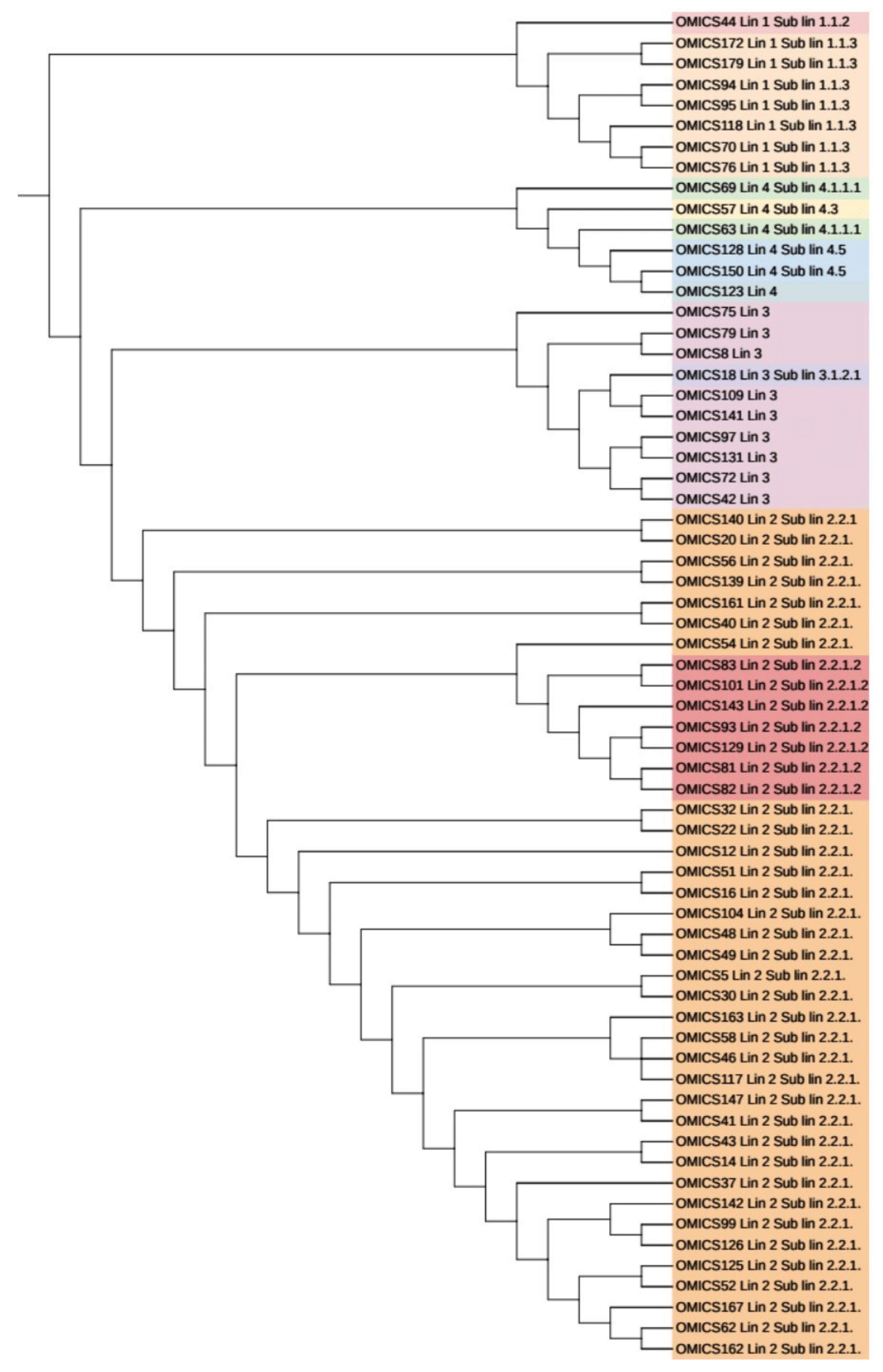
3.6. Phylogenetic Analysis and Identification of Lineages Based on cgMLST and SNP Barcoding
3.7. Phylogenetic Analysis Based on PrincipalGenetic Group (PGG)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2020; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- National Tuberculosis Elimination Programme. India TB Report 2020; National Tuberculosis Elimination Programme: New Delhi, India, 2020. [Google Scholar]
- Lange, C.; Dheda, K.; Chesov, D.; Mandalakas, A.M.; Udwadia, Z.; Horsburgh, C.R. Management of drug-resistant tuberculosis. Lancet 2019, 394, 953–966. [Google Scholar] [CrossRef]
- Husain, A.A.; Kupz, A.; Kashyap, R.S. Controlling the drug-resistant tuberculosis epidemic in India: Challenges and impli-cations. Epidemiol. Health 2021, 43, e2021022. [Google Scholar] [CrossRef] [PubMed]
- Singh, S. Early detection of multi-drug resistant tuberculosis in India using GenoType MTBDRplus assay & profile of resistance mutations in Mycobacterium tuberculosis. Indian J. Med Res. 2014, 140, 477–479. [Google Scholar]
- Gao, Q.; Kripke, K.E.; Saldanha, A.J.; Yan, W.; Holmes, S.; Small, P.M. Gene expression diversity among Mycobacterium tuber-culosis clinical isolates. Microbiology 2005, 151, 5–14. [Google Scholar] [CrossRef]
- Anh, D.D.; Borgdorff, M.W.; Van, L.N.; Lan, N.T.; van Gorkom, T.; Kremer, K.; van Soolingen, D. Mycobacterium tuberculosis Beijing genotype emerging in Vietnam. Emerg. Infect. Dis. 2000, 6, 302–305. [Google Scholar] [PubMed]
- Kato-Maeda, M.; Bifani, P.J.; Kreiswirth, B.N.; Small, P.M. The nature and consequence of genetic variability within Mycobacterium tuberculosis. J. Clin. Invest. 2001, 107, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Mekonnen, A.; Merker, M.; Collins, J.; Addise, D.; Aseffa, A.; Petros, B.; Ameni, G.; Niemann, S. Molecular epidemiology and drug resistance patterns of Mycobacterium tuberculosis complex isolates from university students and the local community in Eastern Ethiopia. PLoS ONE 2018, 13, e0198054. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.; Yusim, K.; Ioerger, T.; Feng, S.; Chase, M.; Greene, M.; Korber, B.; Fortune, S. Mycobacterium tuberculosis—Heterogeneity revealed through whole genome sequencing. Tuberculosis 2012, 92, 194–201. [Google Scholar] [CrossRef]
- Cohen, K.A.; Manson, A.L.; Desjardins, C.A.; Abeel, T.; Earl, A.M. Deciphering drug resistance in Mycobacterium tuberculosis using whole-genome sequencing: Progress, promise, and challenges. Genome Med. 2019, 11, 45. [Google Scholar] [CrossRef]
- Gagneux, S.; DeRiemer, K.; Van, T.; Kato-Maeda, M.; de Jong, B.; Narayanan, S.; Nicol, M.; Niemann, S.; Kremer, K.; Gutierrez, M.C.; et al. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2006, 103, 2869–2873. [Google Scholar] [CrossRef]
- Kohl, T.A.; Diel, R.; Harmsen, D.; Rothgänger, J.; Walter, K.M.; Merker, M.; Weniger, T.; Niemann, S. Whole-genome-based My-cobacterium tuberculosis surveillance: A standardized, portable, and expandable approach. J. Clin. Microbiol. 2014, 5, 2479–2486. [Google Scholar] [CrossRef]
- Jones, R.C.; Harris, L.G.; Morgan, S.; Ruddy, M.C.; Perry, M.; Williams, R.; Humphrey, T.; Temple, M.; Davies, A.P. Phylogenetic Analysis of Mycobacterium tuberculosis Strains in Wales by Use of Core Genome Multilocus Sequence Typing to Analyze Whole-Genome Sequencing Data. J. Clin. Microbiol. 2019, 57, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rufai, S.B.; Singh, A.; Kumar, P.; Singh, J.; Singh, S. Performance of Xpert MTB/RIF Assay in Diagnosis of Pleural Tuberculosis by Use of Pleural Fluid Samples. J. Clin. Microbiol. 2015, 53, 3636–3638. [Google Scholar] [CrossRef]
- Gopinath, K.; Singh, S. Multiplex PCR assay for simultaneous detection and differentiation of Mycobacterium tuberculosis, Mycobacterium avium complexes and other Mycobacterial species directly from clinical specimens. J. Appl. Microbiol. 2009, 107, 425–435. [Google Scholar] [CrossRef]
- Advani, J.; Verma, R.; Chatterjee, O.; Pachouri, P.K.; Upadhyay, P.; Singh, R.; Yadav, J.; Naaz, F.; Ravikumar, R.; Buggi, S.; et al. Whole Genome Sequencing of Mycobacterium tuberculosis Clinical Isolates from India Reveals Genetic Heterogeneity and Region-Specific Variations That Might Affect Drug Susceptibility. Front. Microbiol. 2019, 10, 309. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, S.; Ahmed, A.; Asif, S.; Behera, D.; Javaid, M.; Jani, J.; Jyoti, A.; Mahatre, R.; Mahto, D.; Richter, E.; et al. Direct drug susceptibility testing of Mycobacterium tuberculosis for rapid detection of multidrug re-sistance using the Bactec MGIT 960 system: A multicenter study. J. Clin. Microbiol. 2012, 50, 435–440. [Google Scholar] [CrossRef]
- Rufai, S.B.; Singh, J.; Kumar, P.; Mathur, P.; Singh, S. Association of gyrA and rrs gene mutations detected by MTBDRsl V1 on Mycobacterium tuberculosis strains of diverse genetic background from India. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Technical Manual for Drug Susceptibility Testing of Medicines Used in the Treatment of Tuberculosis; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Kim, H.; Seo, M.; Kil Park, Y.; Yoo, J.-I.; Lee, Y.S.; Chung, G.T.; Ryoo, S. Evaluation of MGIT 960 System for the Second-Line Drugs Susceptibility Testing of Mycobacterium tuberculosis. Tuberc. Res. Treat. 2013, 2013, 1–6. [Google Scholar] [CrossRef]
- Somerville, W.; Thibert, L.; Schwartzman, K.; Behr, M.A. Extraction of Mycobacterium tuberculosis DNA: A Question of Containment. J. Clin. Microbiol. 2005, 43, 2996–2997. [Google Scholar] [CrossRef] [PubMed]
- Black, P.A.; de Vos, M.; Louw, G.E.; van der Merwe, R.G.; Dippenaar, A.; Streicher, E.M.; Abdallah, A.M.; Sampson, S.L.; Victor, T.C.; Dolby, T.; et al. Whole genome sequencing reveals genomic heterogeneity and antibiotic purification in Mycobacterium tuberculosis isolates. BMC Genom. 2013, 16, 857. [Google Scholar] [CrossRef][Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Cingolani, P.; Patel, V.M.; Coon, M.; Nguyen, T.; Land, S.J.; Ruden, D.M.; Lu, X. Using Drosophila melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Front. Genet. 2012, 3, 35. [Google Scholar] [CrossRef]
- Sreevatsan, S.; Pan, X.; Stockbauer, K.E.; Connell, N.D.; Kreiswirth, B.N.; Whittam, T.S.; Musser, J.M. Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proc. Natl. Acad. Sci. USA 1997, 94, 9869–9874. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Silva, M.; Machado, M.P.; Silva, D.N.; Rossi, M.; Moran-Gilad, J.; Santos, S.; Ramirez, M.; Carriço, J.A. chewBBACA: A complete suite for gene-by-gene schema creation and strain identification. Microb. Genom. 2018, 4, e000166. [Google Scholar] [CrossRef]
- Francisco, A.P.; Vaz, C.; Monteiro, P.T.; Melo-Cristino, J.; Ramirez, M.; Carriço, J.A. PHYLOViZ: Phylogenetic inference and data visualization for sequence based typing methods. BMC Bioinform. 2012, 13, 87. [Google Scholar] [CrossRef]
- Chatterjee, S.; Poonawala, H.; Jain, Y. Drug-resistant tuberculosis: Is India ready for the challenge? BMJ Glob. Health 2018, 3, e000971. [Google Scholar] [CrossRef] [PubMed]
- Singhal, R.; Myneedu, V.P.; Arora, J.; Singh, N.; Sah, G.C.; Sarin, R. Detection of multi-drug resistance & characterization of mutations in Mycobacterium tuberculosis isolates from North- Eastern States of India using GenoType MTBDRplus assay. Indian J. Med. Res. 2014, 140, 501–506. [Google Scholar] [PubMed]
- Roetzer, A.; Diel, R.; Kohl, T.A.; Rückert, C.; Nübel, U.; Blom, J.; Wirth, T.; Jaenicke, S.; Schuback, S.; Rüsch-Gerdes, S.; et al. Whole Genome Sequencing versus Traditional Genotyping for Investigation of a Mycobacterium tuberculosis Outbreak: A Longitudinal Molecular Epidemiological Study. PLoS Med. 2013, 10, e1001387. [Google Scholar] [CrossRef]
- Chizimu, J.Y.; Solo, E.S.; Bwalya, P.; Kapalamula, T.F.; Akapelwa, M.L.; Lungu, P.; Shrestha, D.; Fukushima, Y.; Mukonka, V.; Thapa, J.; et al. Genetic Diversity and Transmission of Multidrug-Resistant Mycobacterium tuberculosis strains in Lusaka, Zambia. Int. J. Infect. Dis. 2021, 114, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Coll, F.; McNerney, R.; Guerra-Assunção, J.A.; Glynn, J.R.; Perdigão, J.; Viveiros, M.; Portugal, I.; Pain, A.; Martin, N.; Clark, T.G. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat. Commun. 2014, 5, 4812. [Google Scholar] [CrossRef]
- Kohl, T.A.; Harmsen, D.; Rothgänger, J.; Walker, T.; Diel, R.; Niemann, S. Harmonized Genome Wide Typing of Tubercle Bacilli Using a Web-Based Gene-By-Gene Nomenclature System. EBioMedicine 2018, 34, 131–138. [Google Scholar] [CrossRef]
- Bergval, I.; Sengstake, S.; Brankova, N.; Levterova, V.; Abadía, E.; Tadumaze, N.; Bablishvili, N.; Akhalaia, M.; Tuin, K.; Schuitema, A.; et al. Combined species identification, genotyping, and drug resistance detection of Mycobacterium tuberculosis cultures by MLPA on a bead-based array. PLoS ONE 2012, 7, e43240. [Google Scholar] [CrossRef]
- Nieto Ramirez, L.M.; Ferro, B.E.; Diaz, G.; Anthony, R.M.; de Beer, J.; van Soolingen, D. Genetic profiling of Mycobacterium tuberculosis revealed “modern” Beijing strains linked to MDR-TB from Southwestern Colombia. PLoS ONE 2020, 15, e0224908. [Google Scholar] [CrossRef]
- Rufai, S.B.; Sankar, M.M.; Singh, J.; Singh, S. Predominance of Beijing lineage among pre-extensively drug-resistant and ex-tensively drug-resistant strains of Mycobacterium tuberculosis: A tertiary care center experience. Int. J. Mycobacteriol. 2016, 5 (Suppl 1), S197–S198. [Google Scholar] [CrossRef]
- Gupta, A.; Sinha, P.; Nema, V.; Gupta, P.K.; Chakraborty, P.; Kulkarni, S.; Rastogi, N.; Anupurba, S. Detection of Beijing strains of MDR M. tuberculosis and their association with drug resistance mutations in katG, rpoB, and embB genes. BMC Infect. Dis. 2020, 20, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jiang, X.; Li, W.; Zhang, X.; Wang, W.; Li, C. The study on the association between Beijing genotype family and drug susceptibility phenotypes of Mycobacterium tuberculosis in Beijing. Sci. Rep. 2017, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- San, L.L.; Aye, K.S.; Oo, N.A.T.; Shwe, M.M.; Fukushima, Y.; Gordon, S.; Suzuki, Y.; Nakajima, C. Insight into multidrug-resistant Beijing genotype Mycobacterium tuberculosis isolates in Myanmar. Int. J. Infect. Dis. 2018, 76, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Holt, K.E.; McAdam, P.; Thai, P.V.K.; Thuong, N.T.T.; Ha, D.T.M.; Lan, N.N.; Lan, N.H.; Nhu, N.T.Q.; Hai, H.T.; Ha, V.T.N.; et al. Frequent transmission of the Mycobacterium tuberculosis Beijing lineage and positive selection for the EsxW Beijing variant in Vietnam. Nat. Genet. 2018, 50, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Ajawatanawong, P.; Yanai, H.; Smittipat, N.; Disratthakit, A.; Yamada, N.; Miyahara, R.; Nedsuwan, S.; Imasanguan, W.; Kantipong, P.; Chaiyasirinroje, B.; et al. A novel Ancestral Beijing sublineage of Mycobacterium tuberculosis suggests the transition site to Modern Beijing sublineages. Sci. Rep. 2019, 9, 13718. [Google Scholar] [CrossRef]
- Hanekom, M.; van Pittius, N.G.; McEvoy, C.; Victor, T.; Van Helden, P.; Warren, R. Mycobacterium tuberculosis Beijing genotype: A template for success. Tuberculosis 2011, 91, 510–523. [Google Scholar] [CrossRef]
- Devi, K.R.; Bhutia, R.; Bhowmick, S.; Mukherjee, K.; Mahanta, J.; Narain, K. Genetic Diversity of Mycobacterium tuberculosis Isolates from Assam, India: Dominance of Beijing Family and Discovery of Two New Clades Related to CAS1_Delhi and EAI Family Based on Spoligotyping and MIRU-VNTR Typing. PLoS ONE 2015, 10, e0145860. [Google Scholar] [CrossRef][Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SIRE Drug Susceptibility Pattern by MGIT 960 | Second Line Drug Susceptibility Pattern by MGIT 960 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| STR | INH | RIF | EMB | n | % | AMK | MFX | LFX | LNZ | n | % |
| R | R | R | R | 37 | 29.6% | S | R | R | S | 13 | 35.1% |
| R | R | R | S | 2 | 5.4% | ||||||
| S | R | S | S | 2 | 5.4% | ||||||
| S | S | S | S | 20 | 54.1% | ||||||
| S | R | R | S | 15 | 12.0% | S | R | R | S | 2 | 13.3% |
| S | S | S | S | 13 | 86.7% | ||||||
| S | R | R | R | 5 | 4.0% | S | R | R | S | 2 | 40.0% |
| S | S | S | S | 3 | 60.0% | ||||||
| R | R | R | S | 9 | 7.2% | S | R | R | S | 1 | 11.1% |
| S | S | S | S | 8 | 88.9% | ||||||
| S | S | R | S | 3 | 2.4% | S | S | S | S | 3 | 100.% |
| S | R | S | S | 8 | 6.4% | − | − | − | − | − | − |
| S | S | R | R | 1 | 0.8% | S | S | S | S | 1 | 100% |
| R | S | R | S | 1 | 0.8% | S | S | S | S | 1 | 100% |
| R | R | S | R | 2 | 1.6% | − | − | − | − | − | − |
| R | S | S | S | 1 | 0.8% | − | − | − | − | − | − |
| S | S | S | R | 2 | 1.6% | − | − | − | − | − | − |
| R | R | S | S | 1 | 0.8% | − | − | − | − | − | − |
| S | S | S | S | 40 | 32.0% | − | − | − | − | − | − |
| STRS = 74 STRR = 51 | INHS = 48 INHR = 77 | RIFS = 54 RIFR = 71 | EMBs = 78 EMBR = 47 | n = 125 | AMKS = 69 AMKR =2 | MFXS = 49 MFXR = 22 | LFXS =51 LFXR = 20 | LNZS = 71 | n = 71 | ||
| Drug | Gene | Phenotypic DST | Whole Genome Sequencing Results | |||||
|---|---|---|---|---|---|---|---|---|
| MGIT Result | Mutation | No of Isolates | Lineages | |||||
| Lin1 | Lin2 | Lin3 | Lin4 | |||||
| RIF (n = 41) | rpoB | R | Ser531Leu | 21/41 (51.2%) | 4 (19%) | 15 (71.4) | 2) | − |
| R | Leu511Pro Phe505Leu * | 3/41 (7.3%) | − | 3 (100%) | − | − | ||
| R | Leu511Pro His526Gln | 1/41 (2.4%) | − | 1 (100%) | − | − | ||
| R | Leu511Pro His526Gln Phe505Leu * | 2/41 (4.9%) | − | 2 (100%) | − | − | ||
| R | Asp516Val | 2/41 (4.9%) | − | 1 (50%) | − | − (50%) | ||
| R | Asp516Tyr | 1/41 (2.4%) | − | 1 (100%) | − | − | ||
| R | His526Tyr | 3 /41(7.3%) | 1 (33.3%) | 2 (66.7%) | − | − | ||
| R | His526Asp | 3/41 (7.3%) | - | 3 (100%) | − | − | ||
| R | Gln513Pro | 1/41 (2.4%) | − | − | 1 (100%) | |||
| rpoB rpoC | R | Ser531Leu Ile561Val * Ile572Thr | 1/41 (2.4%) | − | 1 (100%) | − | − | |
| rpoB rpoA | R | Ser531Leu Gly112Ser | 1/41 (2.4%) | 1 (100%) | − | − | − | |
| rpoB rpoA | R | Ser531Leu Gly319Lys | 1/41 (2.4%) | − | 1 (100%) | − | − | |
| rpoB rpoA | R | Ser531Leu Val264Gly | 1/41 (2.4%) | − | − | − | 1 (100%) | |
| rpoB | S | Leu545Met * | 1/41 (2.4%) | − | 1 (100%) | − | − | |
| INH (n = 45) | KatG | R | Ser315Thr | 35/45 (77.8%) | 3 (8.6%) | 27 (77.1%) | 3 (8.6%) | 2 (5.8%) |
| R | Ser450Leu | 1/45 (2.2%) | 1 (100%) | − | − | − | ||
| inhA | R | Ser94Ala | 2/45 (4.4%) | 2 (100%) | − | − | − | |
| ahp | R | 52C>T | 2/45 (4.4%) | 2 (100%) | − | − | − | |
| Fab | R | 15C>T | 1/45 (2.2%) | 1 (100%) | − | − | ||
| Kat Fab | R | Ser140Gly 15C>T | 1/45 (2.2%) | 1 (100%) | − | − | ||
| R | Ser315Thr 17C>T | 1/45 (2.2%) | 1 (100%) | − | − | |||
| R | Ser315Thr 15C>T | 2/45 (4.4%) | 2 (100%) | − | − | |||
| EMB (n = 30) | embB | R | Met306Val | 18/30 (60.0%) | − | 16 (88.8%) | − | 2 (12.2%) |
| R | Gly406Asp | 1/30 (3.3%) | − | − | 1 (100%) | − | ||
| R | Met306Ile | 4/30 (13.3%) | 1 (25%) | 3 (75%) | − | |||
| R | Asp354Ala | 1/30 (3.3%) | − | 1 (100%) | − | − | ||
| R | Met306Leu | 1/30 (3.3%) | − | − | 1 (100%) | − | ||
| embB | R | Glu405Asp | 1/30 (3.3%) | − | 1 (100%) | − | − | |
| R | Gln853ProMet306Val | 2/30 (6.7%) | − | 2 (100%) | − | − | ||
| embA embA | R | Met306Val 12C>T | 2/30 (6.7%) | − | 2 (100%) | − | − | |
| STR (n = 26) | rpsL | R | Lys43Arg | 22/26 (84.6%) | − | 21 (95.4%) | 1 (19.1%) | − |
| R | Lys88Arg | 2/26 (7.7%) | − | 2 (100%) | − | − | ||
| rrs | R | 514 A>C | 2/26 (7.7%) | − | 2 (100%) | − | − | |
| FQ (n = 16) | gyrA | R | Asp94Gly | 6/16 (37.5%) | − | 5 (83.3%) | 1 (16.6%) | − |
| R | Asp94Tyr | 2/16 (12.5%) | − | 2 (100%) | − | − | ||
| R | Asp94Asn | 2/16 (12.5%) | − | 2 (100%) | − | − | ||
| R | Asp94Ala | 1/16 (6.3%) | − | 1 (100%) | − | − | ||
| R | Ala90Val | 1/16 (6.3%) | − | − | − | − (100%) | ||
| R | Asp94His | 1/16 (6.3%) | − | 1 (100%) | − | − | ||
| gyrB | R | Ile486Leu | 1/16 (6.3%) | − | 1 (100%) | − | − | |
| R | Asp461His | 1/16 (6.3%) | − | − | − | 1 (100%) | ||
| R | Ala504Val | 1/16 (6.3%) | − | − | − | − | ||
| PZA (n = 10) | pncA | R | Asp49Ala | 5/10 (50.0%) | − | 5 (100%) | − | − |
| R | Gly108Arg | 2/10 (20.0%) | − | 2 (100%) | − | − | ||
| R | 11A>G | 2/10 (20.0%) | − | 2 (100%) | − | − | ||
| R | Asp136Tyr | 1/10 (10.0%) | 1 (100%) | − | − | − | ||
| AMK (n = 2) | rrs | R | 1484 G>T | 1/2 (50.0%) | − | 1 (100%) | − | − |
| R | 1401 A>G | 1/2 (50.0%) | − | 1 (100%) | − | − | ||
| ETH (n = 8) | inha | NA | Ser94Ala | 2/8 (25.0%) | 2 (100%) | − | − | − |
| fab | NA | 15C>T | 3/8 (37.5%) | − | 3 (100%) | − | − | |
| NA | 17G>T | 1/8 (12.5%) | − | − | − | 1 (100%) | ||
| ethA | NA | 886_886del | 2/8(25.0%) | − | 1 (50%) | − | 1 (50%) | |
| Cysr (n = 2) | alr | NA | Met343Thr | 2/2 (100.0%) | − | 2 (100%) | − | − |
| PAS (n = 2) | thy | NA | 16C>T | 1/2 (50.0%) | − | 1 (100%) | − | − |
| folC | NA | Ile43Thr | 1/2 (50.0%) | − | 1 (100%) | − | − | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mudliar, S.k.R.; Kulsum, U.; Rufai, S.B.; Umpo, M.; Nyori, M.; Singh, S. Snapshot of Mycobacterium tuberculosis Phylogenetics from an Indian State of Arunachal Pradesh Bordering China. Genes 2022, 13, 263. https://doi.org/10.3390/genes13020263
Mudliar SkR, Kulsum U, Rufai SB, Umpo M, Nyori M, Singh S. Snapshot of Mycobacterium tuberculosis Phylogenetics from an Indian State of Arunachal Pradesh Bordering China. Genes. 2022; 13(2):263. https://doi.org/10.3390/genes13020263
Chicago/Turabian StyleMudliar, Shiv kumar Rashmi, Umay Kulsum, Syed Beenish Rufai, Mika Umpo, Moi Nyori, and Sarman Singh. 2022. "Snapshot of Mycobacterium tuberculosis Phylogenetics from an Indian State of Arunachal Pradesh Bordering China" Genes 13, no. 2: 263. https://doi.org/10.3390/genes13020263
APA StyleMudliar, S. k. R., Kulsum, U., Rufai, S. B., Umpo, M., Nyori, M., & Singh, S. (2022). Snapshot of Mycobacterium tuberculosis Phylogenetics from an Indian State of Arunachal Pradesh Bordering China. Genes, 13(2), 263. https://doi.org/10.3390/genes13020263