Abstract
Natural antisense transcripts (NATs) have been generally reported as negative regulators of their sense counterparts. Multidrug and toxic compound extrusion (MATE) proteins mediate the transport of various substrates. Although MATEs have been identified genome-wide in various plant species, their transcript regulators remain unclear. Here, using the publicly available strand-specific RNA-seq datasets of Glycine soja (wild soybean) which have the data from various tissues including developing pods, developing seeds, embryos, cotyledons and hypocotyls, roots, apical buds, stems, and flowers, we identified 35 antisense transcripts of MATEs from 28 gene loci after transcriptome assembly. Spearman correlation coefficients suggested the positive expression correlations of eight MATE antisense and sense transcript pairs. By aligning the identified transcripts with the reference genome of Glycine max (cultivated soybean), the MATE antisense and sense transcript pairs were identified. Using soybean C08 (Glycine max), in developing pods and seeds, the positive correlations between MATE antisense and sense transcript pairs were shown by RT-qPCR. These findings suggest that soybean antisense transcripts are not necessarily negative transcription regulators of their sense counterparts. This study enhances the existing knowledge on the transcription regulation of MATE transporters by uncovering the previously unknown MATE antisense transcripts and their potential synergetic effects on sense transcripts.
1. Introduction
Multidrug and toxic compound extrusion (MATE) transporters typically consist of 12 transmembrane domains (TMDs) [1] and play important roles in cellular transport, metabolism, and physiology [2,3,4]. MATEs are ancient proteins that can be found in all three domains of life [3,5]. Previous research suggested that prokaryotic MATE transporters use H+ or Na+ in exchange for their substrates such as ions and secondary metabolites while most eukaryotic MATE transporters use H+ in exchange for their target substrates [3,5]. In plants, MATE transporters have been reported to be involved in the transportation of various substrates, including ion chelators, phytohormones, alkaloids and flavonoids [4,6,7]. In terms of biological processes, MATE proteins have been reported to regulate various processes such as detoxification, stress tolerance, growth and development, and senescence of plants [8,9]. The diverse substrate specificities of MATE proteins to transport various substrates and regulate diverse biological processes could be the possible reason rendering the big families of MATEs in plant genomes. In a genome-wide survey based on the presence of the conserved MATE domain, 117 MATE genes were identified in the soybean genome [10]. Similarly, 49 MATE genes were identified in Zea mays [11], 67 in Solanum lycopersicum [12], 71 in Populus trichocarpa [13], 53 in Oryza sativa [14], 56 in Arabidopsis thaliana [15], 40 in Medicago truncatula [16], and 65 in Vitis vinifera [17]. Although MATE proteins are structurally similar, as characterized by the typical 12 TMDs [1], they have different substrate specificities [4,6,7], probably due to different substrate recognitions by different amino acid residues [6]. Some substrates transported by MATE proteins are specific to particular groups of plants. For example, anthocyanins are especially rich in the skin of berries and colored seed coats while isoflavones are especially rich the seeds of legumes [18]. VvMATE1 and VvMATE2 were reported to be putative proanthocyanidin transporters in seed berries [19] while GmMATE1 and GmMATE2 were reported to mediate isoflavone transport and storage in soybean seeds [7]. The rich diversity of metabolites in particular plant species hints at the need for a large number of transporters and thus the big MATE families in plants compared to other eukaryotes. Since MATE genes are differentially expressed in different tissue types and different developmental stages [7,20], it is plausible that there are additional regulatory roles that these genes play besides transcriptional regulation.
Natural antisense transcripts (NATs) were believed to negatively regulate their corresponding sense transcripts [21]. For example, in the model plant Arabidopsis, the antisense transcripts covering the entire FLOWERING LOCUS C (FLC locus) were suggested to silence FLC transcripts [22]; the antisense transcript of DOG1 (Delay of Germination 1) was reported to suppress the expression of the sense DOG1 transcript [23]; CDF5 (CYCLING DOF FACTOR 5) transcript and its antisense pair transcript FLORE (CDF5 LONG NONCODING RNA) were reported to inhibit each other’s expression [24]. However, accumulated evidence suggested that NATs are not necessarily the negative regulators of their sense counterparts [25]. For example, also in the model plant Arabidopsis, in a global analysis of antisense and sense transcript pairs that result in elevated small RNA levels, in terms of expression, the transcript pairs could have negative correlations, no correlations, or positive correlations [26]. In another study, MAS, the antisense transcript of MAF4 (MADS AFFECTING FLOWRING4), was reported to be the positive regulator of the transcription of its sense counterpart [27].
The previous soybean NAT database was built using many fewer tissue types [28] or predicted only by genome annotation [29]. Based on the annotation information, transcript orientation, and the degree of overlapping, in the Plant Natural Antisense Transcripts DataBase (PlantNATsDB), it was predicted that there are 436 cis-NATs and 77,903 trans-NATs in soybean [29]. In another genome-wide identification of NATs in soybean, in which sequences of small RNAs were aligned to the predicted cDNA sequences from the soybean gene sequence database, 994 cis-NATs and 25,222 trans-NATs were predicted [28]. However, such predictions relied heavily on the accuracy of transcript annotation [29] and had possibly left out NATs that do not lead to small RNA generation [28].
In this study, we performed a more robust genome-wide analysis to identify NATs that may regulate MATE genes in soybean.
2. Materials and Methods
2.1. Transcriptome Assembly and MATE Antisense Transcripts Identification
The antisense transcript identification was performed with the transcriptome datasets of the wild soybean accessions (Glycine soja) since Glycine soja has more comprehensive strand-specific RNA-seq data from different tissues compared to cultivated soybean (Glycine max) [30,31]. The strand-specific RNA-seq datasets used for transcriptome assembly and MATE antisense transcript identification are summarized in Table S1. Publicly available RNA-seq datasets of Glycine soja (Table S1) were collected. After that, adapter trimming and quality trimming were performed with the raw transcriptome reads using Trimmomatic (0.36) [32]. The clean read data were then aligned to the reference genome of Glycine soja (W05) [31] using HISAT2 (2.1.0) [33], followed by de novo transcriptome assembly using StringTie (1.3.4d) [34]. De novo transcriptome assembly was performed for each sample. After that, the data were merged using the merge function of StringTie [34]. The merged transcriptome data were integrated with the reference annotation of the reference genome of Glycine soja (W05) [31] using the transcript annotation script (NIAP_annotate_v1.1.pl) in the NIAP package (https://github.com/alanlamsiu/NIAP, last accessed on 9 April 2021). This merged transcriptome annotation was then used for MATE antisense transcript identification and expression analysis. Antisense transcripts of MATE loci were identified by intersecting the merged transcriptome annotation and predicted MATE loci for W05 [31] at opposite strands using BEDTools (v2.27.1) [35]. The sequences for these sense and antisense transcripts of MATEs from soybean accession W05 were then extracted. Considering that most research on soybean is done with cultivated soybean (Glycine max), the corresponding MATE antisense and sense transcripts in cultivated soybean were identified with the use of the reference genome of cultivated soybean (Gmax_275_Wm82.a2.v1) [36]. Reciprocal BLAST was performed with all protein sequences of the accession Williams 82 (Wm82) (Gmax_275_Wm82.a2.v1 [36]) and W05 [31] to identify corresponding genes between Wm82 and W05. The transcript sequences of W05 was aligned with the genome sequence of Wm82 (Gmax_275_Wm82.a2.v1 [36]) to identify the corresponding MATE antisense and sense transcripts, which were used later in designing the primers for the RT-qPCR validation experiment. Sequences and coordinates of the W05 and Wm82 antisense transcripts can be found in Supplementary Materials S1 and S2. Genomic loci in W05 corresponding to genes in Wm82 which were annotated as MATE [10] were selected for MATE antisense transcript identification.
2.2. Plant Sample Preparation
Soybean plants (cultivated soybean accession C08 (Glycine max) [37]), were grown on an experimental field in the Chinese University of Hong Kong, watered twice a day, under normal conditions. The developing pods were harvested at 40 days after flowering (DAF). For each biological replicate, the pods were harvested from three individual plants. Two biological replicates were harvested and used for RNA extraction, followed by expression analysis. The pod shells and the seeds with seed coat removed were frozen in liquid nitrogen, then stored at −80 °C before RNA extraction.
2.3. RNA Extraction from Plant Samples
Total RNA was extracted from the frozen pods and seeds without seed coat using Fruit-mate (Cat# 9192, TaKaRa, Shiga, Japan) supplemented with RNasin (recombinant, Cat# N2515, Promega, Madison, WI, USA), and TRIzol Reagent (Cat# 15596018, ThermoFisher Scientific, Waltham, MA, USA) according to the manufacturers’ instructions. The total RNA was treated with DNase I (Cat# 18068015, ThermoFisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol and then used for expression analysis.
2.4. Expression Analysis
The expression levels of the antisense and sense transcripts were validated using One Step PrimeScript RT-PCR Kit (Perfect Real Time) (Cat# RR064B, TaKaRa, Shiga, Japan) according to the manufacturer’s protocol with the following modifications. The reverse primer was added to the DNaseI-treated RNA for reverse transcription. After that, the forward primer was added to the reaction mix for qPCR. The relative expressions of the antisense and sense transcripts were calculated using the 2−ΔΔCT method with VPS as the normalizing gene [38]. Primers used in the experiments are listed in Table S2. Two biological replicates, with each replicate having the total RNA extracted from seeds (seed coats removed) or pods from three individual plants, were used for the expression analysis. Three RT-qPCR reactions were done for each template and primer pair combination. One RT-qPCR was regarded as one technical replicate.
3. Results
3.1. Genome-Wide Identification of MATE Antisense Transcripts
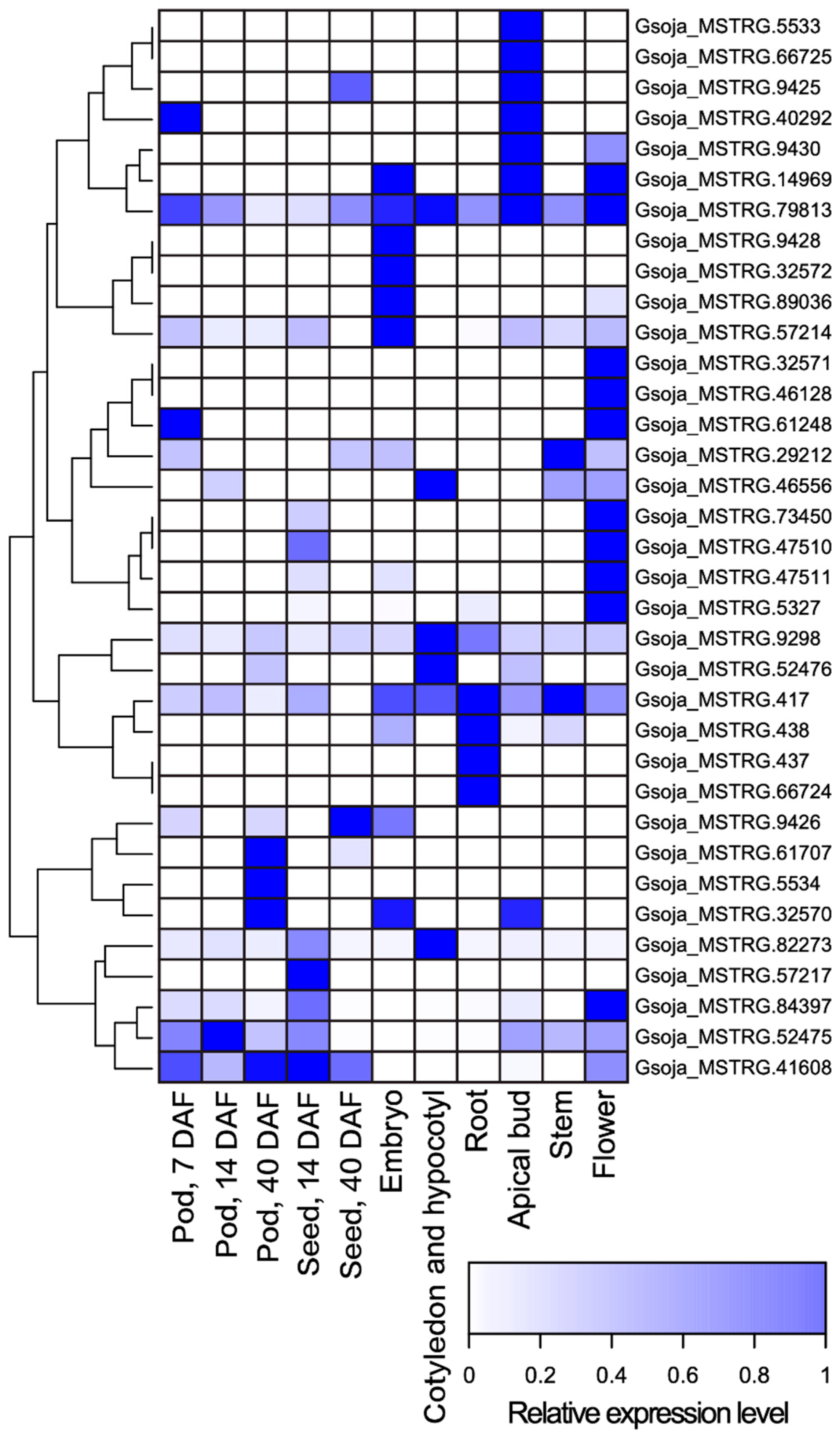
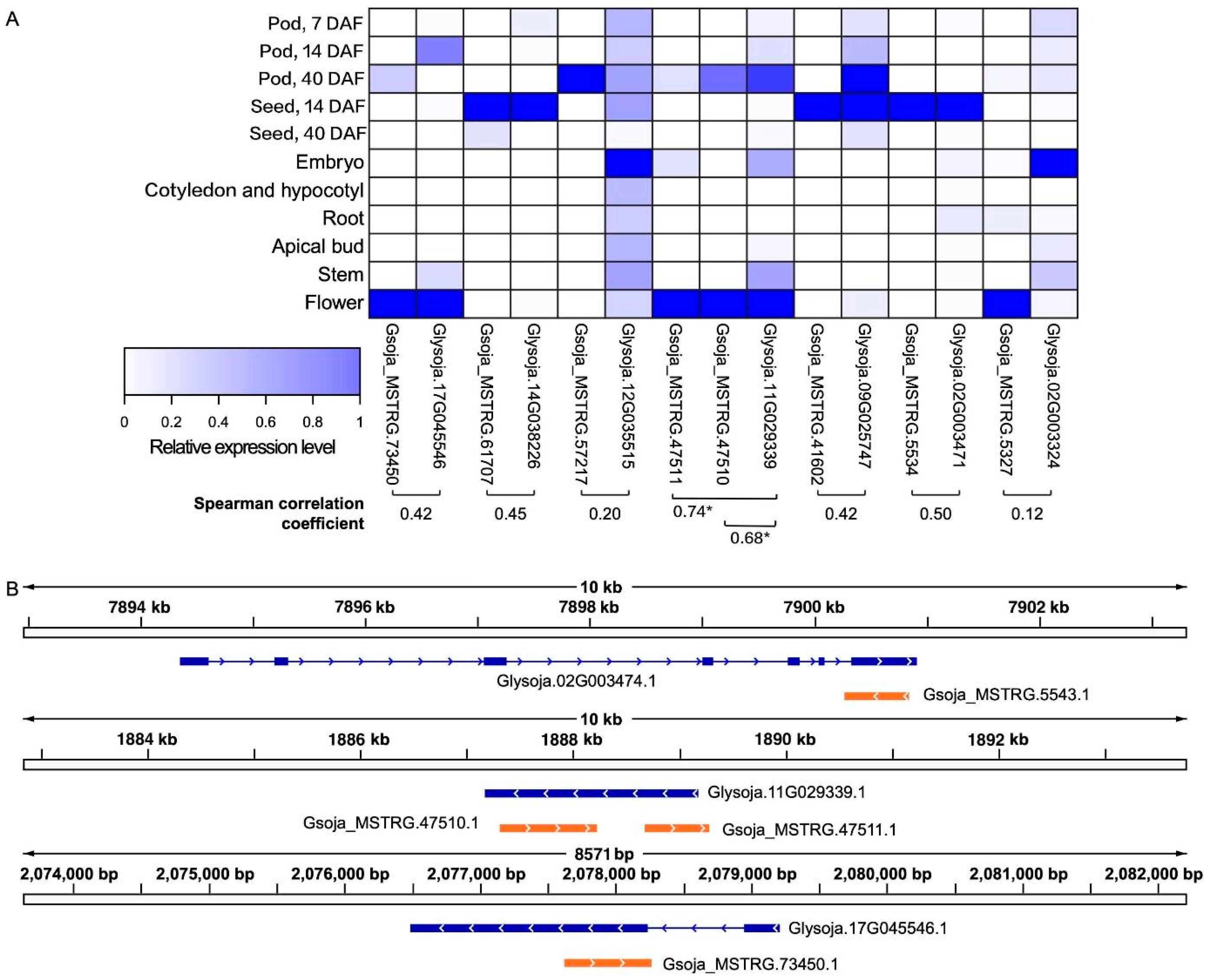
Using the publicly available, strand-specific RNA-seq datasets of a wide variety of tissue types including developing pods, developing seeds, embryos, cotyledons and hypocotyls, roots, apical buds, stems, and flowers, we identified 35 antisense transcripts of MATEs from 28 gene loci in the wild soybean accession W05 (Supplementary Material S1). The predicted relative expression levels of these antisense transcripts are illustrated in Figure 1. Subsequently, we identified the antisense MATE transcripts in the Wm82 by aligning the antisense transcripts identified from strand-specific RNA-seq datasets of W05 to the reference genome of Wm82 (Gmax_275_Wm82.a2.v1 [36]) (Supplementary Material S2).
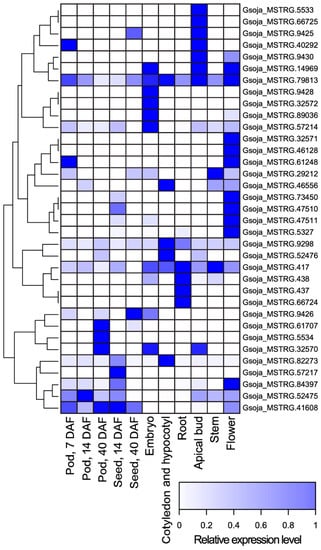
Figure 1.
A heatmap of the predicted relative expression levels of MATE antisense transcripts identified from strand-specific RNA-seq datasets of pod (7 DAF), pod (14 DAF), pod (40 DAF), seed (14 DAF), seed (40 DAF), embryo, cotyledon and hypocotyl, root, apical bud, stem, and flower of soybean accession W05 (Table S1) [30,31].
3.2. Predicted Expression Correlations between MATE Antisense and Their Respective Sense Transcripts
Being NATs, the antisense MATE transcripts could potentially regulate the levels of the respective sense transcripts due to the sequence complementarity. As reflected by the Spearman correlation coefficients, there are several antisense MATE transcripts that exhibited positive expression correlations with their respective sense transcripts in the dataset (Figure 2).
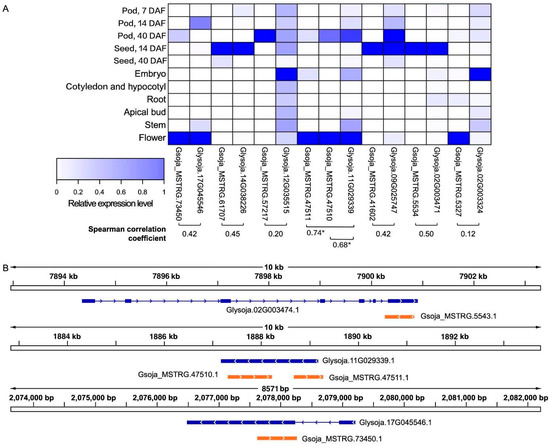
Figure 2.
Antisense MATE transcripts and their sense counterparts in soybean accession W05 with positively correlated expression levels identified based on the publicly available RNA-seq datasets. Left panel. (A) A heatmap of the expression levels of the antisense-sense transcript pairs (joined by a square bracket) that are predicted to be correlated, with the corresponding Spearman correlation coefficients. *Gsoja_MSTRG.47510 and Gsoja_MSTRG.47511 are both antisense transcripts corresponding to the sense transcript Glysoja.11G029339, with the Spearman correlation coefficients being 0.68 and 0.74 respectively; (B) the genomic locations of three of the antisense-sense transcript pairs having high Spearman correlation coefficients are showcased.
3.3. Experimental Validations and Expression Analyses of the Positively Correlated Antisense-Sense MATE Transcript Pairs
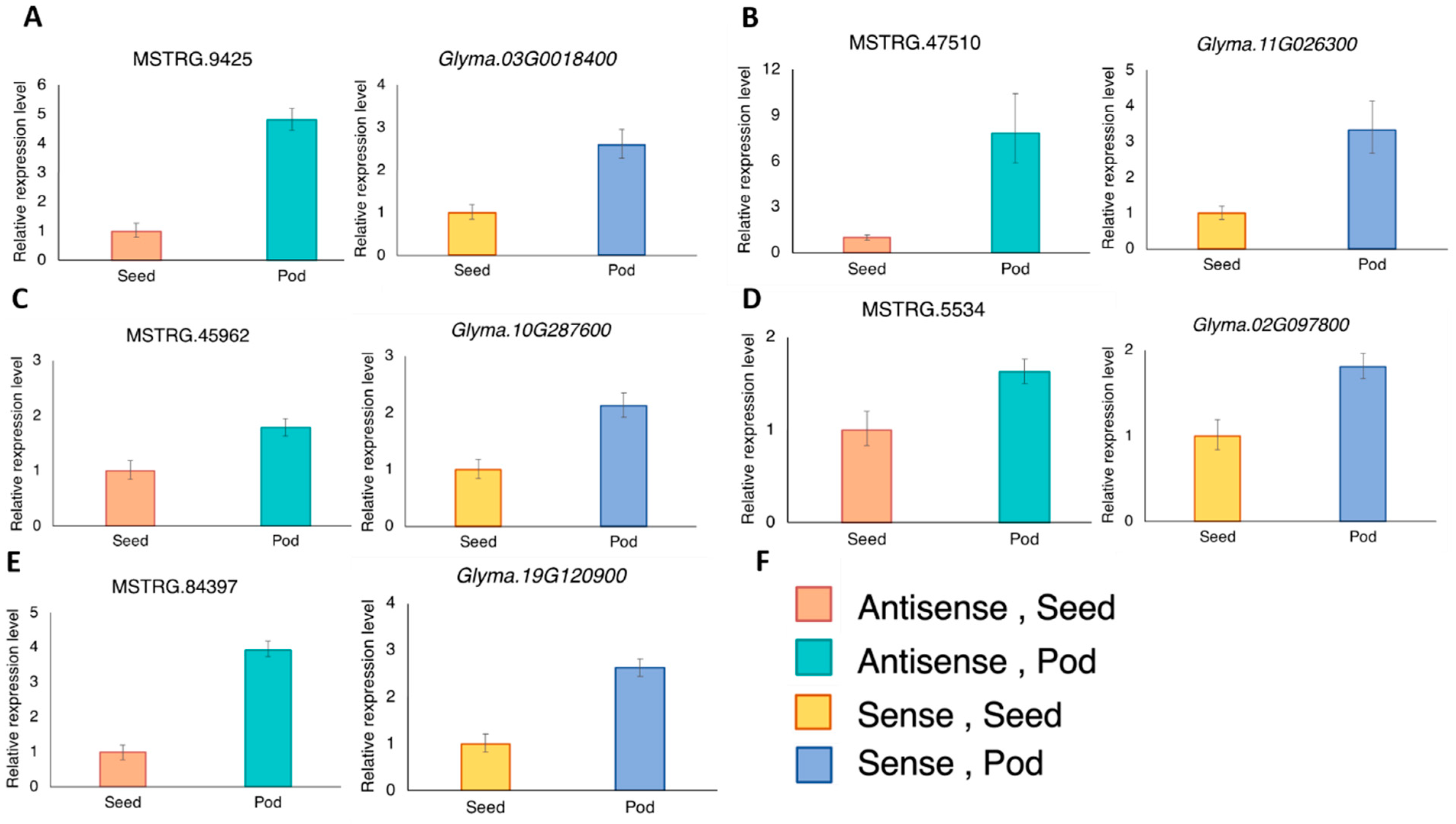
MATE-type proteins are known to be transporters of secondary metabolites [18]. During pod development, nutrients are actively accumulated inside the developing seeds. In this study, developing pods of the soybean accession C08 at 40 DAF, the stage of active seed-filling [39], were used for experimental validation of the transcripts and comparisons of the expression levels between seed and pod (Figure 3). In line with the expression correlations between the antisense and the respective sense transcripts of soybean accession W05 (Figure 2), the antisense and sense MATE transcripts from soybean accession C08 showed similar expression trends between the developing seed and pod (Figure 3).
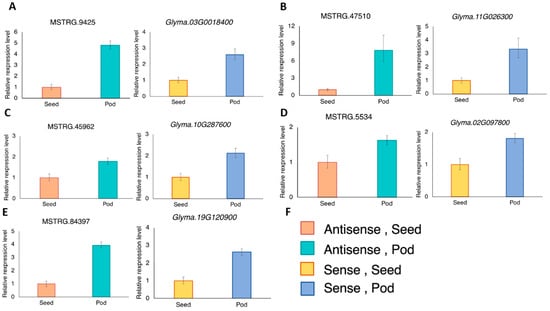
Figure 3.
The expression levels of the antisense and their corresponding sense transcripts in the developing seed and pod were analyzed using RT-qPCR. The antisense and sense pairs are (A) MSTRG.9425 and Glyma03G0018400; (B) MSTRG.47510 and Glyma.11G026300; (C) MSTRG.45962 and Glyma.10G287600; (D) MSTRG.5534 and Glyma.02G097800; (E) MSTRG.84397 and Glyma.19G120900 (GmMATE4 [20]); (F) The color key of the bars shown in panel A–E. Three RT-qPCR reactions were done for each template and primer pair combination. One RT-qPCR was regarded as one technical replicate. Each bar represents the mean of three technical replicates ± standard deviation. The expression was normalized to the reference gene, VPS [38] using the 2−ΔΔCT method. Similar expression trends were obtained from another biological replicate using a separate set of seed and pod from three other plants. Results of the biological replicates are shown in Supplementary Figure S1.
4. Discussion
MATE transporters have diverse physiological roles in growth, development, and stress responses [4,6]. The expression of MATE genes also exhibits differential patterns to cope with their functions [7,13,20]. MATE genes display tissue specific expression patterns. For example, the soybean genes GmMATE1, GmMATE2, and GmMATE4 were shown to have differential expression levels in different tissues such as seeds, seed coats, and pods [7,20]. In Hordeum vulgare, HvAACT1 is expressed in the whole root but has a higher expression level in the root tip [40]. In Nicotiana tabacum, Nt-JAT1 is expressed in leaf, stem, and root [41] while NtJAT2 is expressed in leaf but not stem or root [42]. In terms of the response to stimuli, both Nt-JAT1 and Nt-JAT2 were shown to be induced by methyl jasmonic acid treatment [41,42]. In Populus poplar, PtrMATE1 and PtrMATE2 were shown to be induced by aluminum stress [13]. In Solanum lycopersicu, Sl-ALMT9 was also demonstrated to be induced by aluminum stress [43]. The diverse expression patterns and responses to stimuli of MATE genes imply the possible existence of transcript regulators.
In this study, we identified novel antisense transcripts of MATEs and investigated the expression correlation between NATs and their sense transcripts to reveal the possible negative regulatory roles or the synergetic effects of the NATs on their sense transcripts.
We successfully employed a whole-genome approach to identify high confident NATs of the MATE transcript (Figure 1). These antisense transcripts were not listed in existing databases of soybean antisense transcripts [28,29]. We were able to validate the expressions of the predicted antisense transcripts using RT-qPCR (Figure 3). Among the experimentally validated transcripts (Figure 3), GmMATE4 (Glyma.19G120900, Figure 3E) was previously reported to be localized in the overlapping QTLs regulating the content of antioxidants, phenolics, and flavonoids in soybean seeds [20]. The antisense transcript of GmMATE4 was not reported in the previous study on the expression patterns in developing soybean seeds and pods [20]. The identification of novel MATE antisense transcripts enriches the knowledge of the expression of MATE genes in soybean.
Surprisingly, we found that the relative expression levels of many of the MATE NATs are positively correlated to the corresponding sense transcripts (Figure 2). Whether NATs promote or reduce the target transcript stability has been a subject of debate [25,44]. Nevertheless, there has been accumulating evidence to support the synergy brought forth by cis-NATs on the sense mRNA [25]. In the global identification of Arabidopsis long non-coding RNAs (lncRNAs), based on the Pearson correlation coefficients, the strong tendency of NATs to positively regulate the expression of their sense overlapping genes was suggested [27]. MAS, the NAT overlapping the sense transcript of MADS AFFECTING FLOWERING4 (MAF4), was found to be promoting MAF4 expression at the transcriptional level [27]. It was reported that MAS recruits WDR5a, a core component of COMPASS-like complexes, to enhance histone 3 lysine 4 trimethylation (H3K4me3), which brings forth transcriptional activation of MAF4 [27].
In this study, we predicted positive correlation between antisense transcripts of MATEs and their corresponding sense transcripts based on the Spearman correlation coefficients (Figure 2), and validated the positive correlation of the antisense and sense pairs in developing seeds and pods by RT-qPCR (Figure 3). These results are in line with the possible synergetic effects of the antisense transcripts on the expression of their sense counterparts.
5. Conclusions
In this study, using strand-specific RNA-seq datasets from the tissues of soybean accession W05 and the reference grade genome of W05 [31], we identified a set of MATE antisense transcripts. Based on Spearman correlation coefficients, several antisense transcripts were predicted to have positive correlations with their sense counterparts. By aligning the antisense transcripts identified in the dataset to the accession Williams 82 (Wm82) (Gmax_275_Wm82.a2.v1 [36]), MATE antisense transcripts in Wm82 were identified. The expressions of the antisense transcripts and their sense counterparts were validated in developing soybean seeds and pods. Results showed positively correlated differential expression patterns of the antisense transcripts and their corresponding sense transcripts in both seed and pod. This is consistent with a possible synergistic effect on sense transcript expressions by the antisense counterparts. This study provides new information on the genome-wide identification of MATE genes, demonstrates the enhanced capacity to detect novel antisense transcripts using strand-specific RNA-seq datasets, and enriches our understanding on the possible role of antisense transcripts in transcription regulation.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/genes13020228/s1, Figure S1: The expression levels of the antisense and their corresponding sense transcripts in the developing seed and pod of the second biological replicate were analyzed using RT-qPCR. Supplementary Material S1: Sequences, chromosomal positions, and strand information of MATE antisense transcripts identified in Glycine soja in this study. Supplementary Material S2: Sequences, chromosomal position, and strand information of Glycine max MATE antisense transcripts predicted from the Glycine soja MATE antisense transcripts identified in this study. Table S1: List of strand-specific RNA-seq datasets of Glycine soja used for transcriptome assembly and MATE antisense transcript identification. Table S2: List of primers used in this study.
Author Contributions
Conceptualization, Y.-S.K. and H.-M.L.; methodology, Y.-S.K., X.L., K.F. and T.-F.C.; formal analysis, Y.-S.K., X.L., K.F. and S.-S.C.; investigation, Y.-S.K., X.L., K.F., S.-S.C., T.-F.C. and G.C.; resources, H.-M.L.; data curation, Y.-S.K., X.L. and K.F.; writing—original draft preparation, Y.-S.K., X.L., K.F. and S.-S.C.; writing—review and editing, Y.-S.K. and H.-M.L.; supervision, H.-M.L.; project administration, H.-M.L.; funding acquisition, H.-M.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Hong Kong Research Grants Council General Research Fund (14143916) to H.-M.L.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data generated in this study are available within this manuscript and companion Supplementary Materials.
Acknowledgments
J.Y. Chu copy-edited this manuscript. Any opinions, findings, conclusions or recommendations expressed in this publication do not reflect the views of the Government of the Hong Kong Special Administrative Region or the Innovation and Technology Commission.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shoji, T.; Inai, K.; Yazaki, Y.; Sato, Y.; Takase, H.; Shitan, N.; Yazaki, K.; Goto, Y.; Toyooka, K.; Matsuoka, K.; et al. Multidrug and toxic compound extrusion-type transporters implicated in vacuolar sequestration of nicotine in tobacco roots. Plant Physiol. 2008, 149, 708–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, S.; De Angeli, A.; Fernie, A.R.; Martinoia, E. Intra- and extra-cellular excretion of carboxylates. Trends Plant Sci. 2009, 15, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Omote, H.; Hiasa, M.; Matsumoto, T.; Otsuka, M.; Moriyama, Y. The MATE proteins as fundamental transporters of metabolic and xenobiotic organic cations. Trends Pharmacol. Sci. 2006, 27, 11. [Google Scholar] [CrossRef] [PubMed]
- Takanashi, K.; Shitan, N.; Yazaki, K. The multidrug and toxic compound extrusion (MATE) family in plants. Plant Biotechnol. 2014, 31, 417–430. [Google Scholar] [CrossRef] [Green Version]
- Lu, M. Structures of multidrug and toxic compound extrusion transporters and their mechanistic implications. Channels 2016, 10, 88–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusakizako, T.; Miyauchi, H.; Ishitani, R.; Nureki, O. Structural biology of the multidrug and toxic compound extrusion superfamily transporters. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183154. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.-S.; Ku, Y.-S.; Yung, W.-S.; Cheng, S.-S.; Man, C.-K.; Yang, L.; Song, S.; Chung, G.; Lam, H.-M. MATE-type proteins are responsible for isoflavone transportation and accumulation in soybean seeds. Int. J. Mol. Sci. 2021, 22, 12017. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, N.; Kar, D.; Deepak Mahajan, B.; Nanda, S.; Rahiman, R.; Panchakshari, N.; Bhagavatula, L.; Datta, S. The multitasking abilities of MATE transporters in plants. J. Exp. Bot. 2019, 70, 4643–4656. [Google Scholar] [CrossRef]
- Tang, R.J.; Luan, M.; Wang, C.; Lhamo, D.; Yang, Y.; Zhao, F.-G.; Lan, W.-Z.; Fu, A.-G.; Luan, S. Plant membrane transport research in the post-genomic era. Plant Commun. 2020, 1, 100013. [Google Scholar] [CrossRef]
- Liu, J.; Li, Y.; Wang, W.; Gai, J.; Li, Y. Genome-wide analysis of MATE transporters and expression patterns of a subgroup of MATE genes in response to aluminum toxicity in soybean. BMC Genom. 2016, 17, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Wu, J.; Jiang, Y.; Jin, J.; Zhou, W.; Wang, Y.; Han, G.; Zhao, Y.; Cheng, B. Genomewide analysis of MATE-type gene family in maize reveals microsynteny and their expression patterns under aluminum treatment. J. Genet. 2016, 95, 691–704. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, A.L.; Chaves-Silva, S.; Yang, L.; Maia, L.G.S.; Chalfun-Júnior, A.; Sinharoy, S.; Zhao, J.; Benedito, V.A. Global analysis of the MATE gene family of metabolite transporters in tomato. BMC Plant Biol. 2017, 17, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Meng, H.; Xing, H.; Liang, L.; Zhao, X.; Luo, K. Genome-wide analysis of MATE transporters and molecular characterization of aluminum resistance in Populus. J. Exp. Bot. 2017, 68, 5669–5683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, M.; Sharma, D.; Singh, M.; Tripathi, R.D.; Trivedi, P.K. Expression of OsMATE1 and OsMATE2 alters development, stress responses and pathogen susceptibility in Arabidopsis. Sci. Rep. 2014, 4, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; He, Z.; Pandey, G.K.; Tsuchiya, T.; Luan, S. Functional cloning and characterization of a plant efflux carrier for multidrug and heavy metal detoxification. J. Biol. Chem. 2002, 277, 5360–5368. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Dixon, R.A. MATE transporters facilitate vacuolar uptake of epicatechin 3′-O-glucoside for proanthocyanidin biosynthesis in Medicago truncatula and Arabidopsis. Plant Cell 2009, 21, 2323–2340. [Google Scholar] [CrossRef] [Green Version]
- Gomez, C.; Terrier, N.; Torregrosa, L.; Vialet, S.; Fournier-Level, A.; Verries, C.; Souquet, J.-M.; Mazauric, J.-P.; Klein, M.; Cheynier, V.; et al. Grapevine MATE-type proteins act as vacuolar H+-dependent acylated anthocyanin transporters. Plant Physiol. 2009, 150, 402–415. [Google Scholar] [CrossRef] [Green Version]
- Ku, Y.-S.; Ng, M.-S.; Cheng, S.-S.; Lo, A.W.-Y.; Xiao, Z.; Shin, T.-S.; Chung, G.; Lam, H.-M. Understanding the composition, biosynthesis, accumulation and transport of flavonoids in crops for the promotion of crops as healthy sources of flavonoids for human consumption. Nutrients 2020, 12, 1717. [Google Scholar] [CrossRef]
- Pérez-Díaz, R.; Ryngajllo, M.; Pérez-Díaz, J.; Peña-Cortés, H.; Casaretto, J.A.; González-Villanueva, E.; Ruiz-Lara, S. VvMATE1 and VvMATE2 encode putative proanthocyanidin transporters expressed during berry development in Vitis vinifera L. Plant Cell Rep. 2014, 33, 1147–1159. [Google Scholar] [CrossRef]
- Li, M.W.; Muñoz, N.B.; Wong, C.F.; Wong, F.L.; Wong, K.S.; Wong, J.W.H.; Qi, X.; Li, K.P.; Ng, M.S.; Lam, H.M. QTLs regulating the contents of antioxidants, phenolics, and flavonoids in soybean seeds share a common genomic region. Front. Plant Sci. 2016, 7, 854. [Google Scholar] [CrossRef]
- Vanhée-Brossollet, C.; Vaquero, C. Do natural antisense transcripts make sense in eukaryotes? Gene 1998, 211, 1–9. [Google Scholar] [CrossRef]
- Swiezewski, S.; Liu, F.; Magusin, A.; Dean, C. Cold-induced silencing by long antisense transcripts of an Arabidopsis Polycomb target. Nature 2009, 462, 799–802. [Google Scholar] [CrossRef] [PubMed]
- Fedak, H.; Palusinska, M.; Krzyczmonik, K.; Brzezniak, L.; Yatusevich, R.; Pietras, Z.; Kaczanowski, S.; Swiezewski, S. Control of seed dormancy in Arabidopsis by a cis-acting noncoding antisense transcript. Proc. Natl. Acad. Sci. USA 2016, 113, E7846–E7855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriques, R.; Wang, H.; Liu, J.; Boix, M.; Huang, L.-F.; Chua, N.H. The antiphasic regulatory module comprising CDF5 and its antisense RNA FLORE links the circadian clock to photoperiodic flowering. New Phytol. 2017, 216, 854–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, R.S.; Poirier, Y. Making sense of the natural antisense transcript puzzle. Trends Plant Sci. 2021, 26, 1104–1115. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, B.; Habermann, K.; Arif, M.A.; Weil, H.L.; Garcia-Molina, A.; Kleine, T.; Mühlhaus, T.; Frank, W. Identification of small RNAs during high light acclimation in Arabidopsis thaliana. BMC Plant Biol. 2020, 12, 298. [Google Scholar] [CrossRef]
- Zhao, X.; Li, J.; Lian, B.; Gu, H.; Li, Y.; Qi, Y. Global identification of Arabidopsis lncRNAs reveals the regulation of MAF4 by a natural antisense RNA. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Qiyan, J.; Zhiyong, N.; Hui, Z. Prediction and identification of natural antisense transcripts and their small RNAs in soybean (Glycine max). BMC Genom. 2013, 14, 280. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Yuan, C.; Zhang, J.; Zhang, Z.; Bai, L.; Meng, Y.; Chen, L.-L.; Chen, M. PlantNATsDB: A comprehensive database of plant natural antisense transcripts. Nucleic Acids Res. 2012, 40, D1187–D1193. [Google Scholar] [CrossRef]
- Lin, X.; Lin, W.; Ku, Y.-S.; Wong, F.; Li, M.-W.; Lam, H.-M.; Ngai, S.-M.; Chan, T.-F. Analysis of soybean long non-coding RNAs reveals a subset of small peptide-coding transcripts. Plant Physiol. 2020, 182, 1359–1374. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Chung, C.Y.-L.; Li, M.-W.; Wong, F.-L.; Wang, X.; Liu, A.; Wang, Z.; Leung, A.K.-Y.; Wong, T.-H.; Tong, S.-W.; et al. A reference-grade wild soybean genome. Nat. Commun. 2019, 10, 1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Lam, H.-M.; Xu, X.; Liu, X.; Chen, W.; Yang, G.; Wong, F.-L.; Li, M.-W.; He, W.; Qin, N.; Wang, B. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 2010, 42, 1053–1059. [Google Scholar] [CrossRef]
- Yim, A.K.-Y.; Wong, J.W.-H.; Ku, Y.-S.; Qin, H.; Chan, T.-F.; Lam, H.-M. Using RNA-seq data to evaluate reference genes suitable for gene expression studies in soybean. PLoS ONE 2015, 10, e0136343. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, G.K.; Hajduch, M.; Graham, K.; Thelen, J.J. In-depth investigation of the soybean seed-filling proteome and comparison with a parallel study of rapeseed. Plant Physiol. 2008, 148, 504–518. [Google Scholar] [CrossRef] [Green Version]
- Fujii, M.; Yokosho, K.; Yamaji, N.; Saisho, D.; Yamane, M.; Takahashi, H.; Sato, K.; Nakazono, M.; Ma, J.F. Acquisition of aluminium tolerance by modification of a single gene in barley. Nat. Commun. 2012, 3, 713. [Google Scholar] [CrossRef]
- Morita, M.; Shitan, N.; Sawada, K.; Van Mongtagu, M.C.E.; Inzéc, D.; Rischer, H.; Goossens, A.; Oksman-caldentey, K.; Moriyama, Y.; Yazaki, K. Vacuolar transport of nicotine is mediated by a multidrug and toxic compound extrusion (MATE) transporter in Nicotiana tabacum. Proc. Natl. Acad. Sci. USA 2009, 106, 2447–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shitan, N.; Minami, S.; Morita, M.; Hayashida, M.; Ito, S.; Takanashi, K.; Omote, H.; Moriyama, Y.; Sugiyama, A.; Goossens, A.; et al. Involvement of the leaf-specific multidrug and toxic compound extrusion (MATE) transporter Nt-JAT2 in vacuolar sequestration of nicotine in Nicotiana tabacum. PLoS ONE 2014, 9, e108789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Wang, X.; Hu, T.; Zhang, F.; Wang, B.; Li, C.; Yang, T.; Li, H.; Lu, Y.; Giovannoni, J.J.; et al. An InDel in the promoter of AI-ACTIVATED MALATE TRANSPORTER9 selected during tomato domestication determines fruit malate contents and aluminum tolerance. Plant Cell 2017, 29, 2249–2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brophy, J.A.; Voigt, C.A. Antisense transcription as a tool to tune gene expression. Mol. Syst. Biol. 2016, 12, 854. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).