
An Integrated Linkage Map of Three Recombinant Inbred Populations of Pea (Pisum sativum L.)

 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
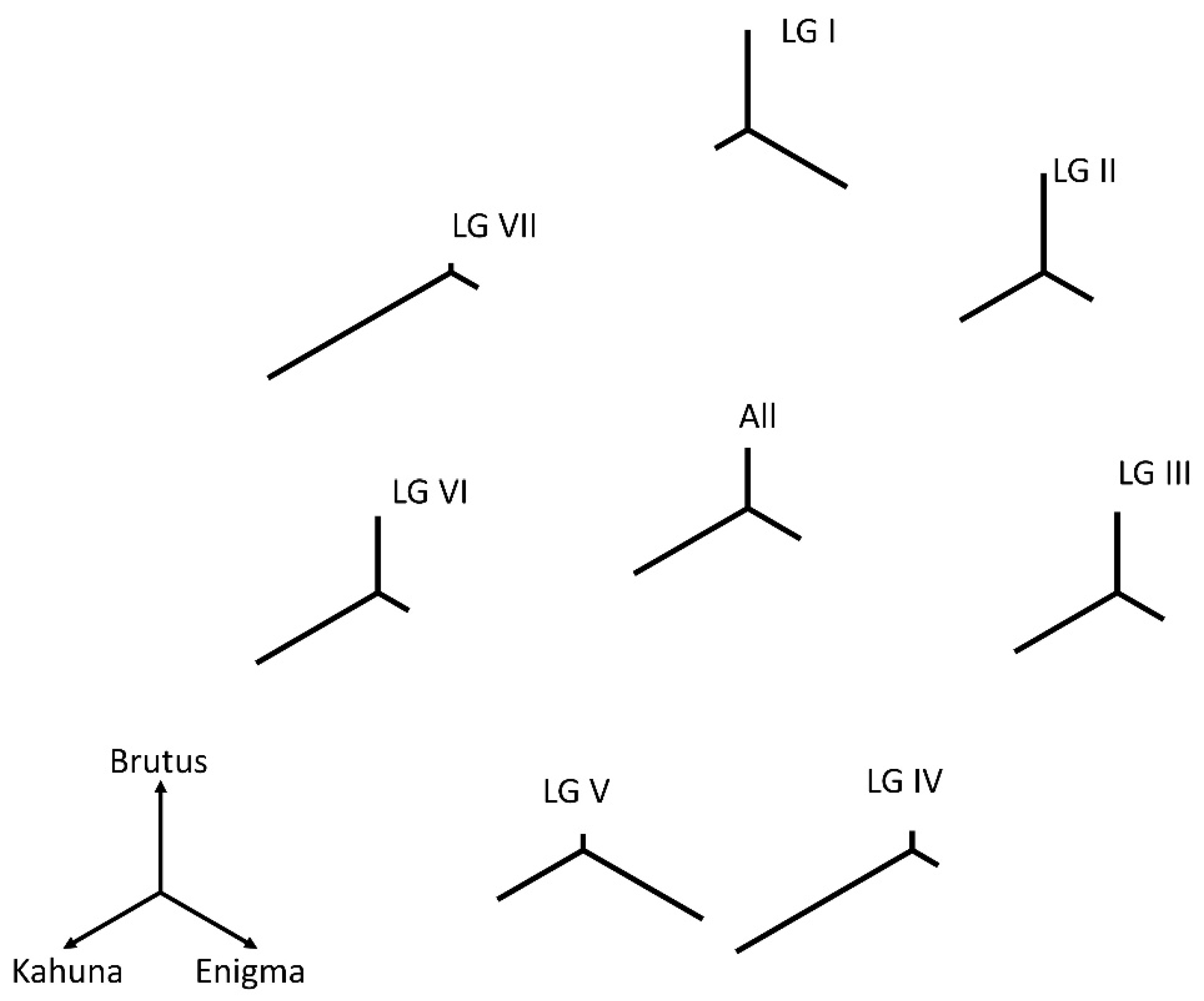
2.1. Plant Material
2.2. SNP Data and Mapping
2.3. Estimates of Relatedness
2.4. Construction of Individual Genetic Maps Using ASMap
2.5. Construction of An Integrated Map
2.6. Single-Marker Analysis of Quantitative Traits
3. Results
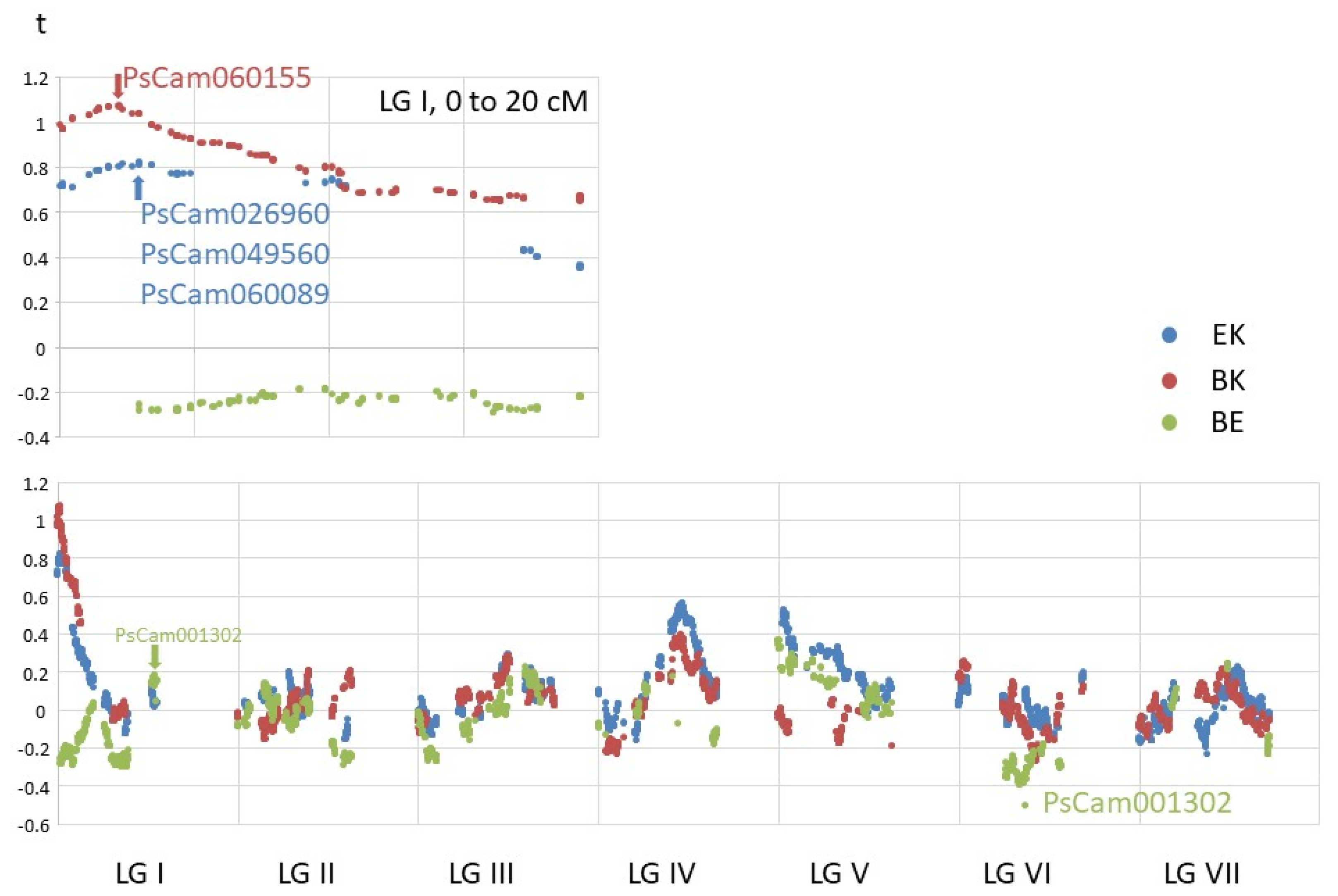
3.1. Relatedness of the Parental Genomes
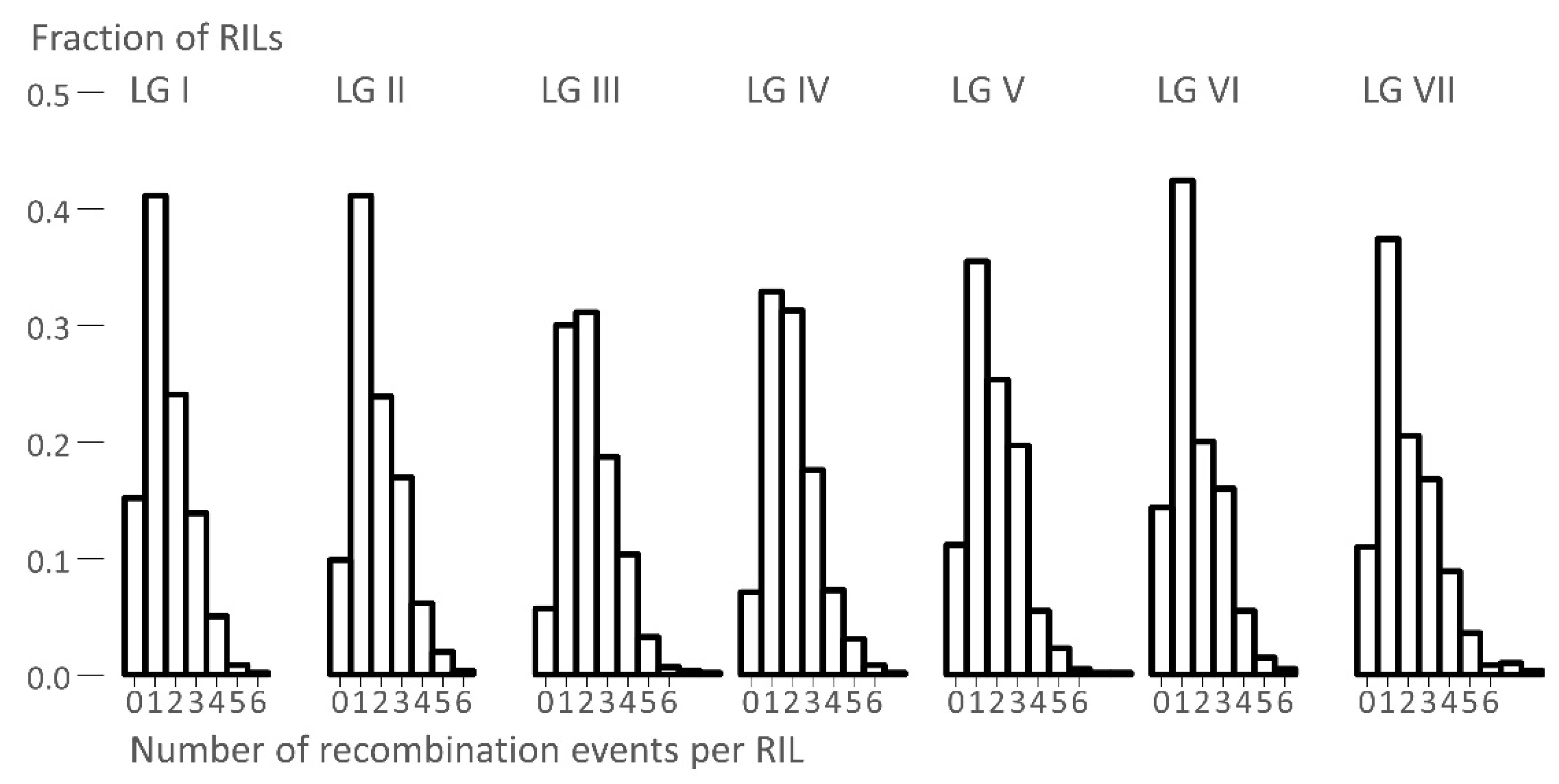
3.2. The Number of Recombination Events Per RIL
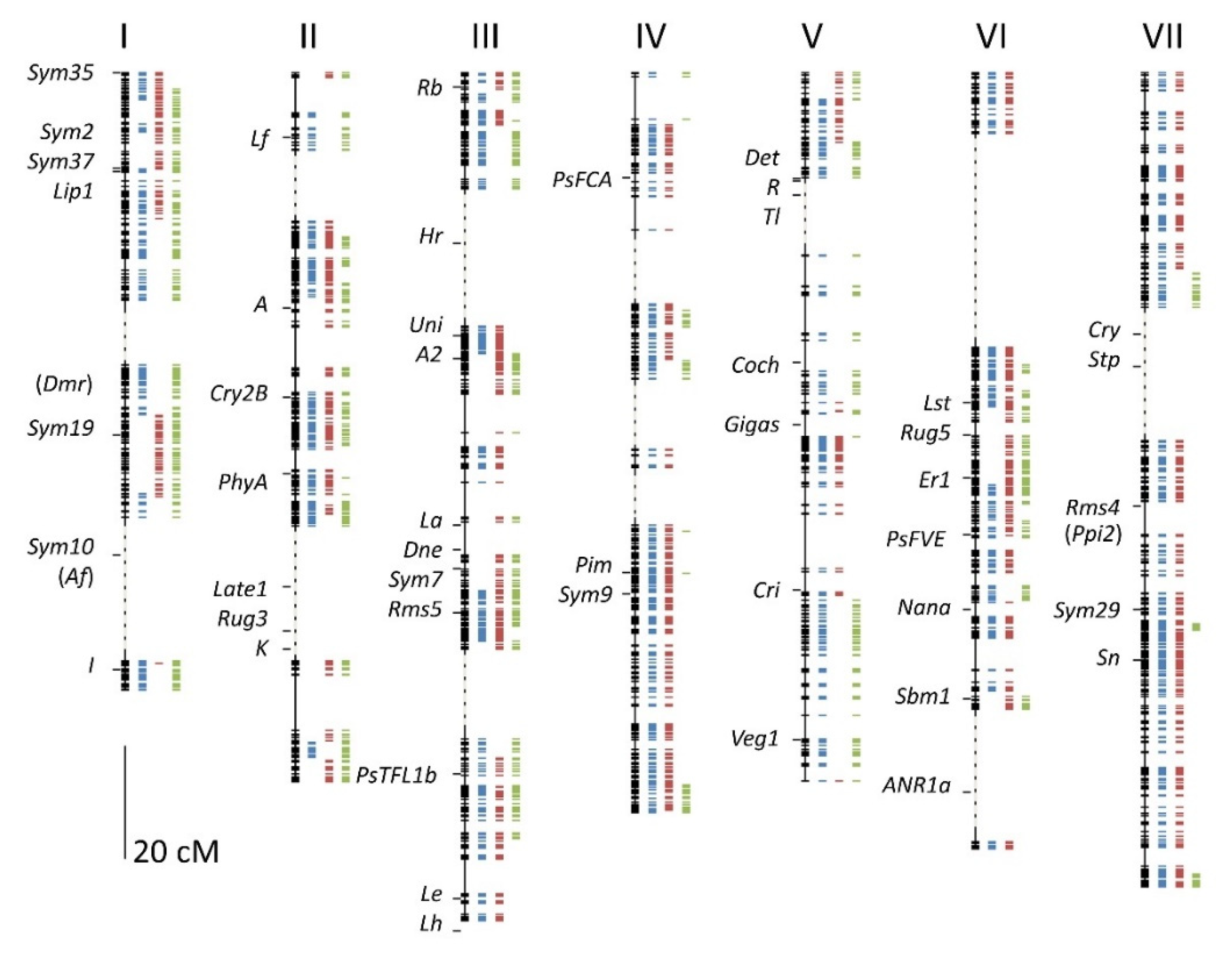
3.3. The Linkage Map
3.4. Recombination: Map Distance between Recombination Events
3.5. Comparison to the Three Individual Genetic Maps
3.6. Genetic and Physical Maps
3.7. Nonlinear Patterns of Alignment
3.8. Quantitative Traits
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
- The frequency distribution of distances between recombination events
- Map distance between recombination events
- Close double recombination events
References
- Moreau, C.; Knox, M.; Turner, L.; Rayner, T.; Thomas, J.; Philpott, H.; Belcher, S.; Fox, K.; Ellis, N.; Domoney, C. Recombinant inbred lines derived from cultivars of pea for understanding the genetic basis of variation in breeders’ traits. Plant Genet. Resour. Charact. Util. 2018, 16, 424–436. [Google Scholar] [CrossRef] [Green Version]
- Kreplak, J.; Madoui, M.-A.; Cápal, P.; Novák, P.; Labadie, K.; Aubert, G.; Bayer, P.E.; Gali, K.K.; Syme, R.A.; Main, D.; et al. A reference genome for pea provides insight into legume genome evolution. Nat. Genet. 2019, 51, 1411–1422. [Google Scholar] [CrossRef] [PubMed]
- Cheema, J.; Ellis, T.H.N.; Dicks, J. THREaD Mapper Studio: A novel, visual web server for the estimation of genetic linkage maps. Nucleic Acid. Res. 2010, 38, W188–W193. [Google Scholar] [CrossRef] [PubMed]
- Tayeh, N.; Aluome, C.; Falque, M.; Jacquin, F.; Klein, A.; Chauveau, A.; Berard, A.; Houtin, H.; Rond, C.; Kreplak, J.; et al. Development of two major resources for pea genomics: The GenoPea 13.2K SNP Array and a high density, high resolution consensus genetic map. Plant J. 2015, 84, 1257–1273. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Bhat, P.R.; Close, T.J.; Lonardi, S. Efficient and Accurate Construction of Genetic Linkage Maps from the Minimum Spanning Tree of a Graph. PLoS Genet. 2008, 4, e1000212. [Google Scholar] [CrossRef] [PubMed]
- Kiss, G.B.; Kereszt, A.; Kiss, P.; Endre, G. Colormapping: A non-mathematical procedure for genetic mapping. Acta Biol. Hung. 1998, 49, 125–142. [Google Scholar]
- Hall, K.J.; Parker, J.S.; Ellis, T.H.N. The relationship between genetic and cytogenetic maps of pea. I. Standard and translocation karyotypes. Genome 1997, 40, 744–754. [Google Scholar] [CrossRef]
- Haldane, J.B.S.; Waddington, C.H. Inbreeding and linkage. Genetics 1931, 16, 357–374. [Google Scholar] [CrossRef]
- Haldane, J.S.B. The combination of linkage values, and the calculation of distance between loci of linked factors. J. Genet. 1919, 8, 299–309. [Google Scholar]
- Hecht, V.; Foucher, F.; Macknight, R.; Vardy, M.E.; Ellis, N.; Rameau, C.; Weller, J.L. Conservation of Arabidopsis flowering genes in model legumes. Plant Physiol. 2005, 127, 1420–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foucher, F.; Morin, J.; Courtiade, J.; Cadioux, S.; Ellis, N.; Banfield, M.J.; Rameau, C. DETERMINATE and LATE FLOWERING are two TERMINAL FLOWER 1/CENTRORADIALIS homologues controlling two distinct phases of flowering initiation and development in pea. Plant Cell 2003, 15, 2742–2754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, J.L.; Hecht, V.; Liew, L.C.; Sussmilch, F.C.; Wenden, B.; Knowles, C.L.; Vander Schoor, J.K. Update on the genetic control of flowering in garden pea. J. Exp. Bot. 2009, 60, 2493–2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, J.L.; Ortega, R. Genetic control of flowering time in legumes. Front. Plant Sci. 2015, 6, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berdnikov, V.A.; Gorel, F.L.; Bogdanova, V.S.; Kosterin, O.E.; Trusov, Y.A.; Rozov, S.M. Effect of a substitution of a short chromosome segment carrying a histone H1 locus on expression of the homeotic gene Tl in heterozygote in the garden pea Pisum sativum L. Genet. Res. 1999, 73, 93–109. [Google Scholar] [CrossRef]
- Zhu, H.; Cannon, S.B.; Young, N.D.; Cook, D.R. Phylogeny and Genomic Organization of the TIR and Non-TIR NBS-LRR Resistance Gene Family in Medicago truncatula. Mol. Plant-Microbe Interact. 2002, 15, 529–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mood, A.M. The Distribution Theory of Runs. Ann. Mat. Stat. 1940, 11, 367–392. [Google Scholar] [CrossRef]
- Hellens, R.; Moreau, C.; Lin-Wang, K.; Schwinn, K.E.; Thomson, S.J.; Fiers, M.W.E.J.; Frew, T.J.; Murray, S.R.; Hofer, J.M.I.; Jacobs, J.M.E.; et al. Identification of Mendel’s White Flower Character. PLoS ONE 2010, 10, e13230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavanagh, C.; Morell, M.; Mackay, I.; Powell, W. From mutations to magic: Resources for gene discovery, validation and delivery in crop plants. Curr. Opin. Plant Biol. 2008, 11, 215–221. [Google Scholar] [CrossRef]
- Hultén, M.A. On the origin of crossover interference: A chromosome oscillatory movement (COM) model. Mol. Cytogenet. 2011, 4, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Marker | SNP | Segregates in | Common | Identity | ||
|---|---|---|---|---|---|---|
| PsCam004787 | T/C | BK | EK | K | B = E | |
| PsCam014211 | T/G | BE | EK | E | B = K | |
| PsCam042135 | T/G | BE | BK | B | E = K | |
| Number of Segregating Markers Scored | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| By Population | Unique to | Shared by | ||||||||||
| Total | EK | BK | EB | EK | BK | EB | EK & BK | EK & EB | BK & EB | |||
| LG I | 550 | 327 | 295 | 461 | 7 | 7 | 3 | 75 | 245 | 213 | ||
| LG II | 726 | 438 | 560 | 444 | 2 | 7 | 1 | 273 | 163 | 280 | ||
| LG III | 975 | 664 | 768 | 518 | 0 | 0 | 0 | 457 | 207 | 311 | ||
| LG IV | 605 | 556 | 530 | 118 | 0 | 4 | 2 | 483 | 73 | 43 | ||
| LG V | 456 | 425 | 206 | 276 | 3 | 1 | 1 | 176 | 246 | 29 | ||
| LG VI | 576 | 398 | 491 | 254 | 2 | 7 | 0 | 313 | 83 | 171 | ||
| LG VII | 668 | 637 | 580 | 107 | 5 | 4 | 3 | 552 | 80 | 24 | ||
| total | 4556 | 3445 | 3430 | 2178 | 19 | 30 | 10 | 2329 | 1097 | 1071 | ||
| Linkage. | Number of Crossovers | |||||||
|---|---|---|---|---|---|---|---|---|
| Group | Mean | Variance | Maximum | Variance/Mean | ||||
| I | 1.56 | 1.26 | 6 | 0.81 | ||||
| II | 1.76 | 1.39 | 6 | 0.79 | ||||
| III | 2.13 | 1.64 | 8 | 0.77 | ||||
| IV | 1.98 | 1.52 | 7 | 0.77 | ||||
| V | 1.83 | 1.57 | 8 | 0.85 | ||||
| VI | 1.62 | 1.44 | 6 | 0.89 | ||||
| VII | 1.96 | 2.14 | 8 | 1.09 | ||||
| Total Length | Number of | cM/Mb | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| LG | cM | Mb | Marker Intervals | Mean | SD | |||||
| I | 110 | 342 | ch2 | 490 | 0.52 | 1.70 | ||||
| II | 126 | 384 | ch6 | 611 | 0.41 | 0.71 | ||||
| III | 151 | 463 | ch5 | 832 | 0.32 | 0.32 | ||||
| IV | 132 | 357 | ch4 | 544 | 0.52 | 2.07 | ||||
| V | 126 | 438 | ch3 | 442 | 0.36 | 0.29 | ||||
| VI | 138 | 298 | ch1 | 508 | 0.40 | 0.76 | ||||
| VII | 145 | 393 | ch7 | 583 | 0.46 | 0.68 | ||||
| All | 928 | 2675 | 4010 | 0.42 | 1.09 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sawada, C.; Moreau, C.; Robinson, G.H.J.; Steuernagel, B.; Wingen, L.U.; Cheema, J.; Sizer-Coverdale, E.; Lloyd, D.; Domoney, C.; Ellis, N. An Integrated Linkage Map of Three Recombinant Inbred Populations of Pea (Pisum sativum L.). Genes 2022, 13, 196. https://doi.org/10.3390/genes13020196
Sawada C, Moreau C, Robinson GHJ, Steuernagel B, Wingen LU, Cheema J, Sizer-Coverdale E, Lloyd D, Domoney C, Ellis N. An Integrated Linkage Map of Three Recombinant Inbred Populations of Pea (Pisum sativum L.). Genes. 2022; 13(2):196. https://doi.org/10.3390/genes13020196
Chicago/Turabian StyleSawada, Chie, Carol Moreau, Gabriel H. J. Robinson, Burkhard Steuernagel, Luzie U. Wingen, Jitender Cheema, Ellen Sizer-Coverdale, David Lloyd, Claire Domoney, and Noel Ellis. 2022. "An Integrated Linkage Map of Three Recombinant Inbred Populations of Pea (Pisum sativum L.)" Genes 13, no. 2: 196. https://doi.org/10.3390/genes13020196
APA StyleSawada, C., Moreau, C., Robinson, G. H. J., Steuernagel, B., Wingen, L. U., Cheema, J., Sizer-Coverdale, E., Lloyd, D., Domoney, C., & Ellis, N. (2022). An Integrated Linkage Map of Three Recombinant Inbred Populations of Pea (Pisum sativum L.). Genes, 13(2), 196. https://doi.org/10.3390/genes13020196