Abstract
In his influential book “Evolution by Gene Duplication”, Ohno postulated that frameshift mutation could lead to a new function after duplication, but frameshift mutation is generally thought to be deleterious, and thus drew little attention in functional innovation in duplicate evolution. To this end, we here report an exhaustive survey of the genomes of human, mouse, zebrafish, and fruit fly. We identified 80 duplicate genes that involved frameshift mutations after duplication. The frameshift mutation preferentially located close to the C-terminus in most cases (55/88), which indicated that a frameshift mutation that changed the reading frame in a small part at the end of a duplicate may likely have contributed to adaptive evolution (e.g., human genes NOTCH2NL and ARHGAP11B) otherwise too deleterious to survive. A few cases (11/80) involved multiple frameshift mutations, exhibiting various patterns of modifications of the reading frame. Functionality of duplicate genes involving frameshift mutations was confirmed by sequence characteristics and expression profile, suggesting a potential role of frameshift mutation in creating functional novelty. We thus showed that genomes have non-negligible numbers of genes that have experienced frameshift mutations following gene duplication. Our results demonstrated the potential importance of frameshift mutations in molecular evolution, as Ohno verbally argued 50 years ago.
1. Introduction
The significance of gene duplication for genetic innovation was first recognized by Stephens in the 1950s [1], and then popularized by Ohno in the 1970s [2]. Ohno postulated that one duplicate could evolve a new function by mutations, the scenario later well recognized as neofunctionalization. The evolution of gene duplication on a genomic scale has been extensively studied in a wide range of species, most of which argued the relative contribution of neofunctionalization and its derivatives, including subfunctionalization, by looking at amino acid evolution by point mutations and/or expression changes [3].
It is interesting to note that, in his seminal book Evolution by Gene Duplication [2], Ohno pointed out a potential role of frameshift mutations in neofunctionalization:
“Starting with a frame-shift mutation, a duplicate might acquire a new function, which is totally different from that assigned to the original gene. Admittedly, this is a one in a million chance, but in evolution, events with the odds of one in one million occurred time and time again”.
We re-recognized the importance of frameshift mutations from recent reports on duplication of the human NOTCH genes [4,5]. After the split with chimpanzee, a copy of human NOTCH2 genes acquired a new function by a 4 bp deletion that introduced a fragment of completely new amino acid sequences. This duplicate, named NOTCH2NL, was further duplicated, and was speculated to have contributed to the rapid evolution of the human brain size. This striking case is exactly what Ohno pointed out, thereby motivating us to explore the contribution of frameshift mutations in the evolution of duplicate genes. Our literature survey found the human ARHGAP11 genes as another convincing case [6,7,8,9,10]. ARHGAP11B arose from duplication of ARHGAP11A in the human lineage after separation from the chimpanzee lineage, and evolved the new function of increasing basal progenitor amplification during neocortex development. The new function of ARHGAP11B is acquired by a 55 deletion in its mRNA that leads to loss of RhoGAP activity by GAP domain truncation, and addition of a human-specific carboxy-terminal amino acid sequence [6,7].
Beside these clear cases, there have been very few documentations of frameshift mutations involved in the adaptive evolution. Several genomic surveys for frameshifts between duplicate genes have been reported [11,12,13,14] in animals. However, most of the identified ones were involved in complex alternative splicing and exon shuffling, and none was a convincing case of Ohno’s prediction, except for one case in the rodent lineage (the functional divergence between NKG2A and NKG2C) [11]. In plants, putative frameshift mutations were found in the MADS-box gene family, the B-function DEF/AP3 subfamily, the A-function SQUA/AP1 subfamily, and the E-function AGL2 subfamily, all of which are involved in the specification of organ identity during flower development [15].
We here report an exhaustive survey of the genomes of human (Homo sapiens), mouse (Mus musculus), zebrafish (Danio rerio), and fruit fly (Drosophila melanogaster) to identify frameshift mutation in duplicates. We evaluate and argue for the contribution of the scenario of duplication–frameshift–neofunctionalization in these four diverse organisms and its evolutionary consequences.
2. Materials and Methods
Genomic data was retrieved from Ensembl (release 88). To identify putative duplicates, TBLASTX [16] of all coding sequences (CDS) against themselves was first performed with an E-value cutoff of 1 × 10−7 in each genome. This meant that all transcripts for each gene were included to exclude frameshifts that were introduced by alternative splicing. Then, high-scoring pairs (HSP) with both overlap and identification ≥ 70% were selected as putative duplicates using custom Perl scripts. Next, a protein sequence from one CDS of a HSP was mapped to another CDS of the HSP with GeneWise [17] for each HSP to identify HSPs with frameshift mutation. HSPs with frameshift mutations were selected as potential candidates. Considering that alternative splicing commonly results in frameshift mutation among different transcripts in a gene, only candidate HSPs in each of which all transcripts from one gene against all transcripts from another gene with consistently the same frameshift mutation were kept. As such, redundancy of the candidate HSPs with frameshifts was also reduced from transcript to gene level. Thirdly, duplication events of the identified duplicated pair with frameshift mutations were confirmed with a comparative genomics profile in Ensembl. Finally, the candidate duplicated pairs with frameshift mutations were manually checked with great care for orthology, synteny, and sequence alignment by utilizing multiple sequence alignments of orthologs and paralogs in Ensembl.
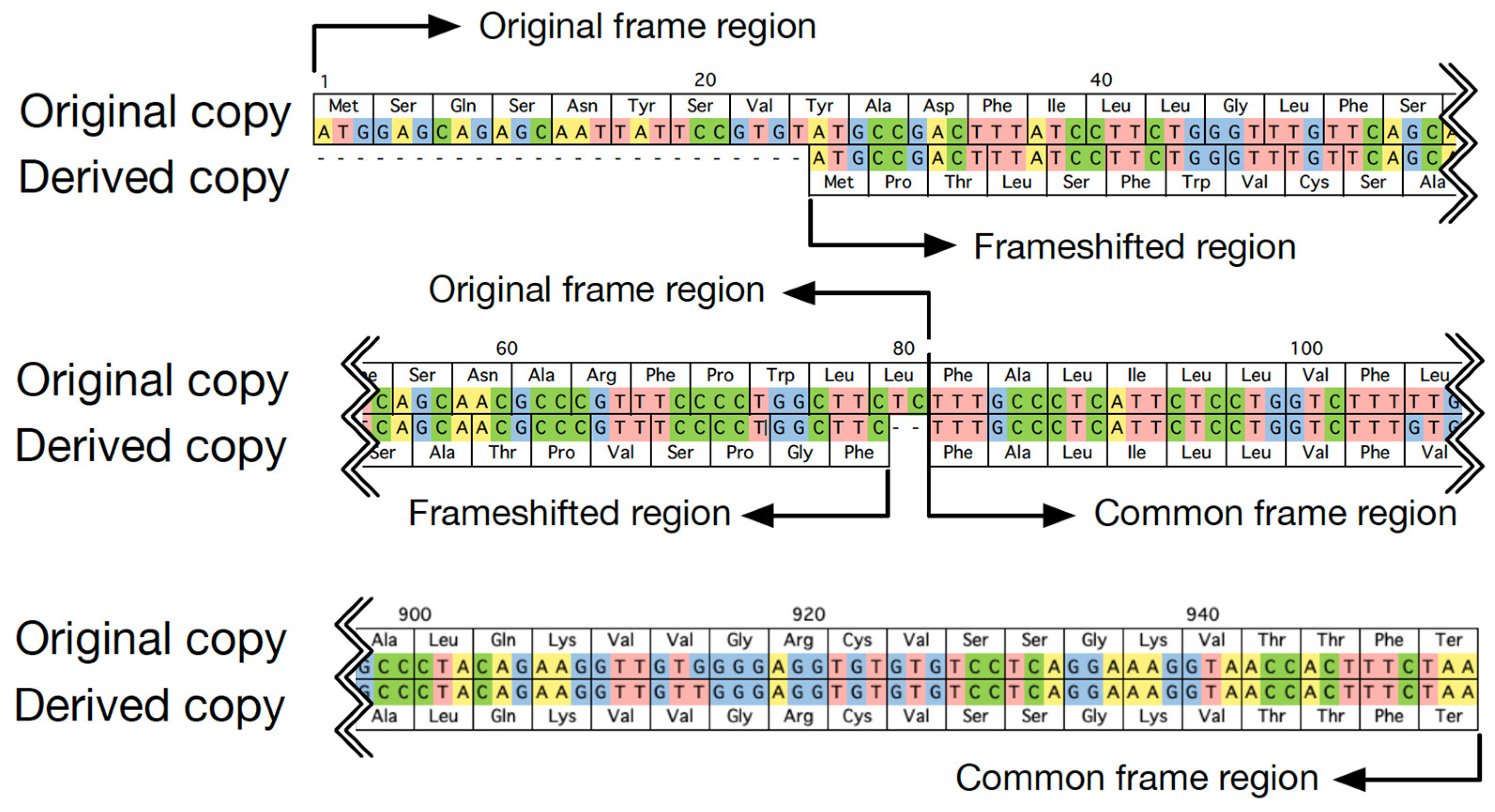
To best investigate the evolutionary trajectory of frameshift mutations in duplicates, frameshift mutations were defined by considering the outgroup; that is, the sequence from the most closely related species free from the duplication event. In cases without available outgroup sequences, frameshift mutations were defined in the shorter copy by considering characteristics of the copy with frameshift mutations in cases with outgroup sequences. First, location and length of frameshift mutations were sketched. An example is shown in Figure 1. The region in which both copies used the same reading frame was referred to as a common frame region. The region using the new reading frame in the derived copy was called the frameshifted region, and its corresponding part in the original copy was called the original frame region. Then, the divergence and selection pressure between duplicates in their common frame and frameshift region were characterized, respectively, by calculating the numbers of synonymous (Ks) and nonsynonymous substitutions (Ka) per site using codeml with the pairwise model in PAML4 [18]. Specifically, to estimate the selection pressure, an ancestry sequence was first reconstructed based on duplicates and their closely related outgroup sequence at the nucleotide level. Then, Ka and Ks in each copy of duplicates were calculated by comparison between the duplicate sequence and its ancestry sequence for the common frame region and the frameshifted region, respectively. For the calculation of Ka and Ks, we used the original reading frame.
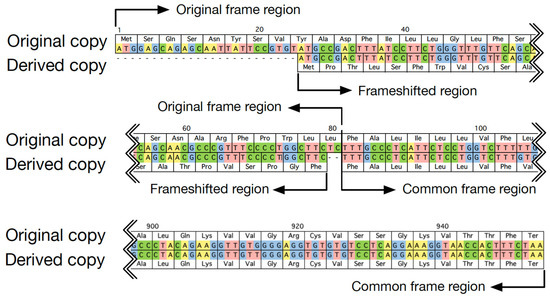
Figure 1.
An example of duplicated genes, with one copy having experienced a frameshift mutation (H10, OR2T7, and OR2T27 in human in Table 1).
The gene expression information was retrieved from GTEx (https://www.gtexportal.org/home/, accessed on 2 April 2020) for human, and from MGI (http://www.informatics.jax.org/, accessed on 2 April 2020) for mouse. The gene expression information for zebrafish was obtained by mapping RNA-seq data from SRA to its genome with HISAT and StringTie pipeline [19]. The statistics were done in R version 3.3.3.
3. Results and Discussion
We identified in total 80 pairs of duplicate genes with frameshift mutations between them in these species (see Materials and Methods): 17 in human (NOTCH2 genes were not found because they were not annotated in Ensembl release 88), 30 in mouse, 31 in zebrafish, and 2 in fruit fly (Table 1). For each pair of duplicates, we searched for an outgroup sequence, which allowed us to determine which copy underwent frameshift mutation(s) (referred to as the derived copy) and which was the original copy. To do so, we computed Ks_C and Ka_C, the numbers of synonymous and nonsynonymous substitutions per site in common frame regions, respectively. We predicted that Ks_C roughly gave the age of the duplication, while knowing the gene conversion between them retarded the divergence [20]. We then searched for an outgroup sequence such that Ks_C was sufficiently smaller than the typical species divergence at synonymous sites. We further looked at the synteny to confirm their orthology. We determined the original and derived copies for 53 cases (Tables S1 and S2).

Table 1.
Duplicated genes with frameshift mutations.
We first looked at the number and the locations of frameshift mutations. We found that most cases (69/80) experienced only one frameshift mutation, while the others underwent multiple frameshift mutations (11/80). The 69 cases with one frameshift mutation were categorized into type-C and -N classes according to whether the frameshift mutation changed the reading frame in the C- or N-terminus of the amino acid sequence. Only a representative case is shown for type-C and -N classes in Figure 2A,B, respectively. We found that the majority was categorized into the type-C class (51 vs. 18, p = 0.0001, binomial test). The enrichment of type-C was reasonable, because it can be explained by a single mutation alone; that is, the reading frame downstream of the mutation was changed by the mutation. In contrast, although only one mutation was detected for a type-N case, it should be noted that another mutation upstream of the start codon is needed to create a type-N (otherwise, it becomes a type-C).
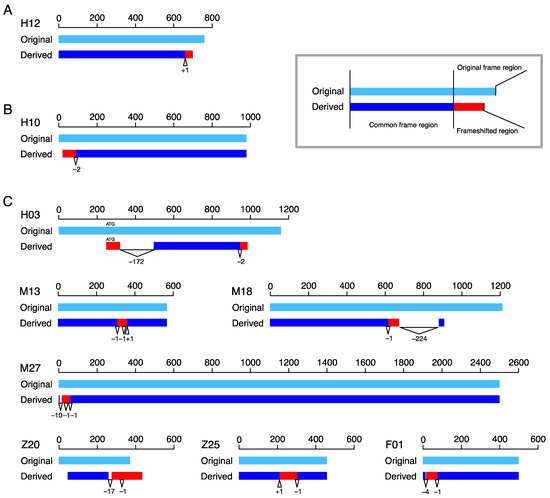
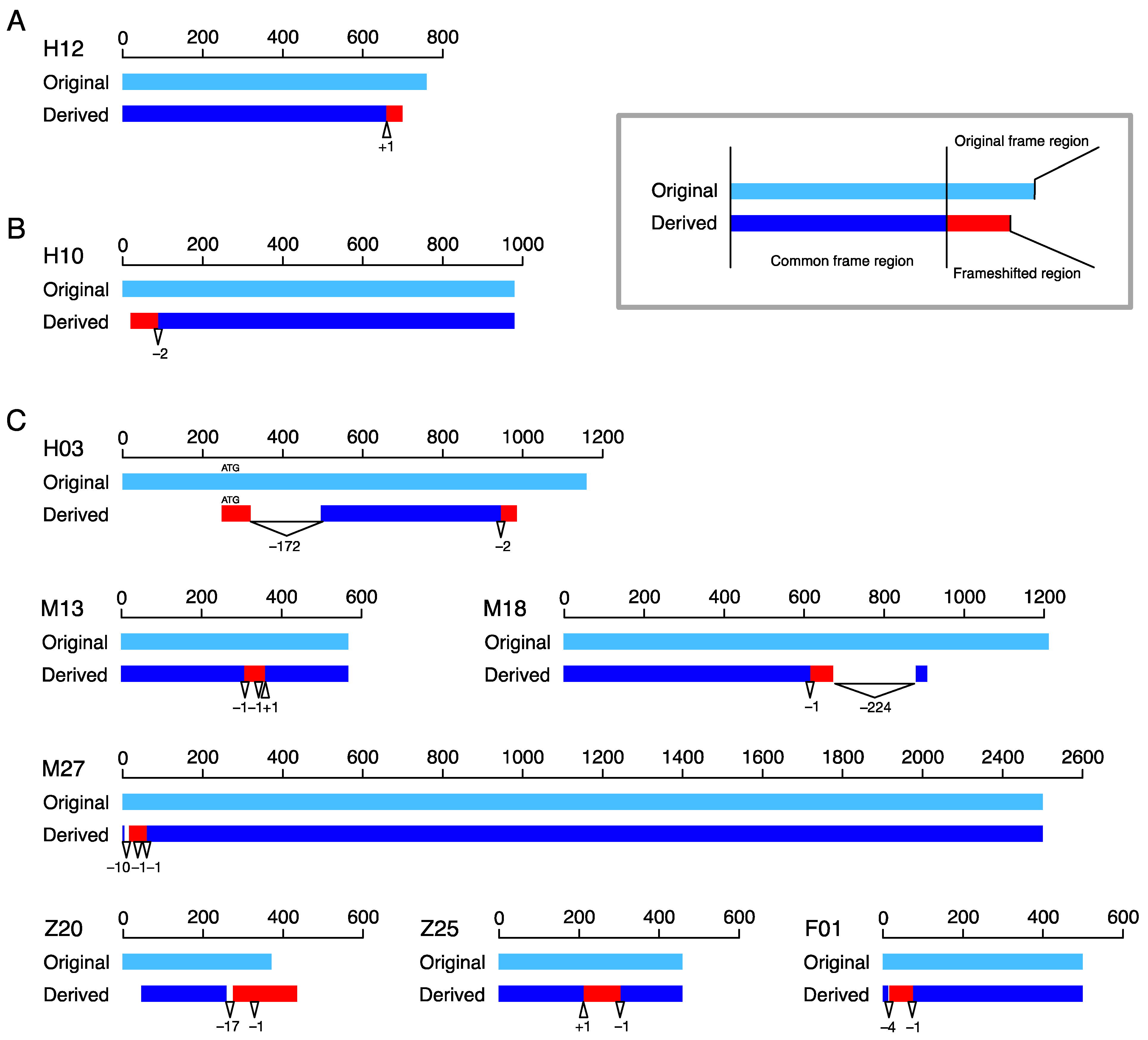
Figure 2.
Locations of typical frameshift mutations along their coding sequences in bp from the start codon. The original and derived copies are presented in light blue and blue, with frameshifted regions in red. Triangles with positive numbers are insertions, and reverse triangles with negative numbers are deletions. The numbers are the sizes of the indels. (A) One representative case for type-C. (B) One representative case for type-N. (C) Seven cases involving multiple frameshift mutations: H12, CASTOR3, and CASTOR2 in human; H10, OR2T7, and OR2T27 in human; H3, MICA, and MICA in human; M13, Rpl-ps1, and Rpl-ps6 in mouse; M18, Lipo5, and Lipo3 in mouse; M27, Pcdhb7, and Pcdhb9 in mouse; Z20 and Z25 in zebrafish (gene names are unavailable); and F01, CSNK2B, and CSNK2B in fruit fly.
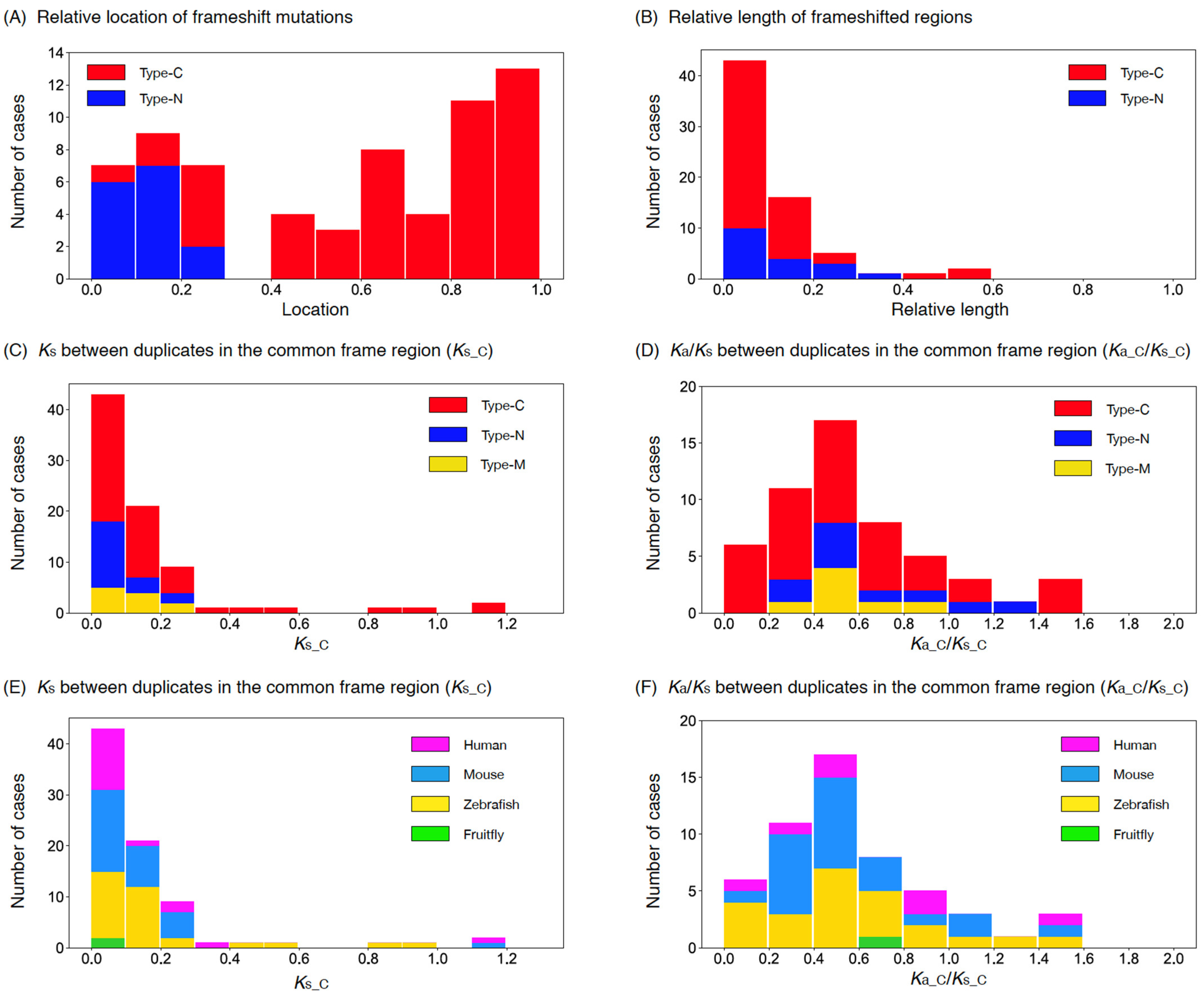
If we used those with an outgroup sequence available (34 type-C and 12 type-N cases), we found that the frameshifted region was shorter than the original frame region in >80% cases (27/34 and 11/12 cases for type-C and -N, respectively). If the entire coding region of the original copy was assigned to the unit interval (0,1), the average relative length of the frameshifted region (0.16 and 0.10 for type-C and -N cases, respectively) was shorter than the original frame region (0.30 and 0.21 for type-C and -N cases, respectively), and the difference was significant (p = 0.05 and 0.01 for type-C and -N cases, respectively; permutation test). It was suggested that a frameshift mutation likely resulted in a shorter coding region, which was also seen in NOTCH2NL [4,5] and ARHGAP11B [6,7].
Figure 3A shows the distribution of relative locations of frameshift mutations, where we used these 46 cases with an outgroup available and also 23 cases with no outgroup (in total, 51 type-C and 18 type-N cases). For the latter cases, given the above results, we assumed that the shorter ones were derived copies, with one exceptional case M22 (Table S1). It was found that their distributions were not uniform, and that most frameshift mutations were found close to the stop codon of the original copy for type-C cases and close to the start codon for type-N cases. Figure 3B shows the distribution of relative length of the frameshifted region, indicating that the length of the frameshifted region in general was shorter than 20% of the entire region.
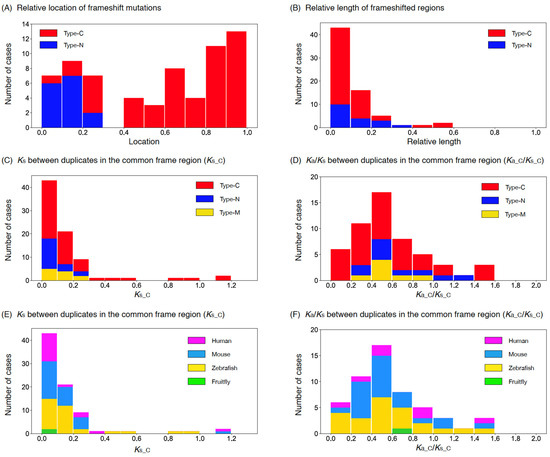
Figure 3.
(A) Distribution of the locations of frameshift mutations mapped on the original copy rescaled to the (0,1) interval. (B) Distribution of the size of frameshifted region relative to the original copy. (C) Distribution of Ks_C for type-C, -N, and -M. (D) Distribution of Ka_C/Ks_C for type-C, -N, and -M. Data for the four species are pooled. (E) Distribution of Ks_C for human, mouse, zebrafish, and fruit fly. (F) Distribution of Ka_C/Ks_C for human, mouse, zebrafish, and fruit fly. Data for type-C, -N, and -M are pooled. The data were binned with a window of size 0.1 in all panels.
The patterns of the 11 cases with multiple frameshift mutations greatly varied; for 7 of these, we obtained an outgroup sequence, and the alignment of the original and derived copies are shown in Figure 2C. The 11 cases included 3 Type-C cases with 2 mutations (Z01, Z18, and Z20, of which Z20 is shown in Figure 2C). These cases were most likely created as follows. A first frameshift mutation created a frameshifted region in the C-terminus, and a second frameshift mutation occurred downstream of the first one, causing another new reading frame. A similar pattern was observed in six cases (M13, M18*, M25, M27*, Z10, and F01*, of which those with a star are shown in Figure 2C), where more than one mutation created a frameshifted region in the middle of the coding region (type-M). For these cases, it is reasonable to consider that a first mutation created a frameshifted region in the C-terminus, and a secondary frameshift mutation occurred downstream of the first one. The difference was that a secondary mutation reversed the reading frame back to the original reading frame. Other complex cases included H03 (Figure 2C), which experienced two frameshift mutations, resulting in two distinct frameshifted regions, one in the C-terminus and the other in the N-terminus.
Figure 3C,D show the distributions of Ks_C and Ka_C/Ks_C for 51 type-C, 18 type-N, and 11 multiple-mutation cases. We found that most cases had Ks_C < 0.3, while some type-C cases had Ks_C > 0.3. The distributions of Ka_C/Ks_C for the three types seemed similar: the majority had Ka_C/Ks_C < 1, with an average of 0.65. The exact same distributions are shown with labels of the four species (Figure 3E,F). In Figure S1, the configurations of common frame and frameshifted regions for all 80 cases are shown.
Provided that the most likely fate of a duplicate is to become a pseudogene, a major question is whether those frameshift mutations played a meaningful role in functional genes, or they were merely random deleterious mutations being accumulated on the way to becoming a pseudogene. To address this question, we first asked if those frameshifted duplicates were functional in the current genome. We looked over the list of the duplicates, and found that most of them were well-annotated genes (78/80; Table 1), including ARHGAP11 (our ID: H08) in human, as mentioned above [6,7]; oocyte-specific homeobox (Obox) genes (our ID: M16) [21] and vomeronasal type-1 receptor (Vmn1r) genes (our ID: M21, 22, and 24) [22] in mouse; and alpha-actinin genes (our ID: Z30) in zebrafish [23]. We also confirmed the presence of expression in 78/80 cases, and all of them showed divergence in the expression amount and/or pattern, further supporting their functional importance (Table 1).
Further evidence for stable preservation of the derived frameshifted copy should be seen in their DNA sequences. First, we found that 37/80 cases had Ks_C > 0.1. A relatively large divergence between paralogs (i.e., Ks_C > 0.1) should indicate that these derived copies had been preserved as functional genes for a significant amount of time, while maintaining a low Ka_C. This meant that they had successfully escaped pseudogenization, because pseudogenization usually occurs in a relatively short time after duplication [3].
Next, if frameshift mutations significantly contributed to the acquisition of novel functions, we predicted we would observe accelerated amino acid substitutions in the derived copy, particularly in the frameshifted region. A testable prediction would be that the Ka/Ks ratio was exceeded on the lineage, leading to the derived copy (denoted by Ka_D/Ks_D), particularly in the frameshifted region (denoted by Ka_DF/Ks_DF). We tested this for 49 cases with a reliable outgroup sequence available (Table S2), for which we estimated the ancestral sequence for each pair of duplicates, and synonymous and nonsynonymous divergences from the ancestral sequence to the original and derived copies were computed (denoted by Ka_O and Ks_O for the original copy and Ka_D and Ks_D for the derived copy). Note that we used the original reading frame for this calculation. We found one case (Z13) that exhibited Ka_D/Ks_D significantly larger than 1 for the entire region (Ka_D = 0.058 vs. Ks_D =0.000, p < 0.03). By focusing on the frameshifted region, we tested the null hypothesis of an equal Ka/Ks ratio for the original and derived lineages (i.e., Ka_OF/Ks_OF = Ka_DF/Ks_DF), and found one case (Z13) with a significant deviation (Ka_OF/Ks_OF = 0.000, Ka_DF/Ks_DF = 2.423, p < 0.0001). One might suspect that the observation might be explained by relaxed selection of the derived copy, but this should not be the case, due to a low Ka/Ks ratio in the common frame region (Ka/Ks = 0.27), indicating that the Ka/Ks ratio was elevated only in the frameshifted region, and selective constraint remained in the common frame region. We were unable to detect statistically significant results for the other cases, mostly due to a small number of substitutions because the frameshifted regions were generally very short (Figure S1). If we pooled all cases, we obtained Ka_OF/Ks_OF = 0.374, Ka_DF/Ks_DF = 1.087, suggesting that the amino acid substitution rate was on average large on the lineage, leading to the derived copy in the frameshifted region (but not in the common frame region: Ka_OC/Ks_OC = 0.449, Ka_DC/Ks_DC = 0.519).
Thus, for the majority of the cases, there were reasons that supported significant functional roles of the derived copies in the current genome. We then argued the implications of these results on the potential roles of frameshift mutations. First, it is interesting to point out that if a frameshift mutation occurred in an intact coding gene, it always resulted in type-C; that is, the reading frame downstream of the mutation was modified. All other types (i.e., types-N, -M, etc.) needed subsequent mutations. This should be the reason why the most frequent type was type-C. In type-C cases, frameshift mutations for the C-type were enriched near the C-terminus. Assuming mutation occurred randomly along a gene, the skewed observed distribution should indicate that frameshift mutations were in general deleterious and selected against, especially when enough occurred to change the majority of the reading frame. The condition that a frameshift mutation directly contributes to adaptive evolution is that it confers a selective advantage by creating a new reading frame. Most likely, it should occur without changing the major part of the coding gene, otherwise it would be too deleterious to survive. If the new reading frame is beneficial, the frameshift mutation may be favored by selection and preserved in the genome. Another possible scenario would be that the new reading frame is nearly neutral, so that it is not immediately selected out. Then, subsequent amino acid changing mutations could occur in the frameshifted region to create a beneficial amino acid sequence. Such a secondary mutation has to occur before the first mutation is selected out.
In a similar vein, other kinds of secondary mutations could confer selective advantages, and their outcomes include type-N and -M. Suppose a first frameshift mutation creates a type-C gene. When a secondary frameshift mutation occurs downstream of the first one, it will become a type-M gene if it changes the downstream reading frame to the original frame (i.e., a frameshifted region in the middle of the coding region is observed). As mentioned earlier, to explain a type-N gene, a secondary mutation is needed to create a new translation start site upstream of the first mutation, by which a type-C gene immediately turns out to be a type-N gene. Provided that we observed all frameshift mutations close to the N-terminus (Figure 2A), one possible scenario was that these first frameshift mutations killed the original function and produced pseudogenes temporarily, and secondary rescue mutations recovered the original reading frame, perhaps quickly, resulting in type-N genes. In this work, we found some cases (2/80) with multiple frameshift mutations that could be explained by this scenario. Considering that the rate of such rescue mutations was very low, our observation might be difficult to explain by neutral evolution alone. In addition, it should be considered that the results of our study critically depended on genomic assembly and annotation quality, and sequencing and gene prediction errors may affect studies such as ours here. We expect that long-read sequencing could be utilized to validate our findings in future studies.
4. Conclusions
We thus demonstrated that genomes have non-negligible numbers of genes that have experienced frameshift mutations following gene duplication. In most cases, the majority of the original amino acid sequence was kept, and a small part was modified. A frameshift mutation produced a random amino acid sequence downstream until a premature stop codon arises. It should be very rare that such a random sequence provides a novel function for neofunctionalization, but on a genomic scale, we observed a number of cases that benefited from frameshift mutations. Most especially, the studies of NOTCH genes [4,5] and ARHGAP11 genes [6,7,8,9,10] in human suggest that the functional innovation through gene duplication followed by frameshift mutation could play an important role in adaptive evolution of functional traits. Our results demonstrated the potentially importance of frameshift mutations in molecular evolution, as Ohno verbally argued 50 years ago.
Supplementary Materials
The following are available online at www.mdpi.com/article/10.3390/genes13020190/s1, Figure S1: Relative location of the common frame region and frameshifted region in the derived copy; Table S1: Summary of duplicated genes with frameshift mutations; Table S2: Summary of sequence analyses.
Author Contributions
Conceptualization, B.G. and H.I.; methodology, M.Z., H.I., T.S. and B.G.; software, M.Z. and T.S.; validation, M.Z., T.S., B.G. and H.I.; formal analysis, M.Z., T.S., B.G. and H.I.; resources, B.G.; data curation, M.Z., T.S., B.G. and H.I.; writing—original draft preparation, B.G. and H.I.; writing—review and editing, B.G. and H.I.; visualization, T.S. and H.I.; funding acquisition, B.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (grant nos. 32022009 and 31970382) and the Chinese Academy of Sciences (ZDBS-LY-SM005 and the Pioneer Hundred Talents Program).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data supporting reported results can be found in the Supplementary Materials.
Acknowledgments
We also thank Manyuan Long and three anonymous reviewers for thoughtful and constructive comments on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Stephens, S.G. Possible Significance of Duplication in Evolution. In Advances in Genetics Incorporating Molecular Genetic Medicine; Academic Press: Cambridge, MA, USA, 1951; Volume 4, pp. 247–265. [Google Scholar]
- Ohno, S. Evolution by Gene Duplication; Springer: Berlin, Germany, 1970. [Google Scholar]
- Innan, H.; Kondrashov, F. The evolution of gene duplications: Classifying and distinguishing between models. Nat. Rev. Genet. 2010, 11, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Fiddes, I.T.; Lodewijk, G.A.; Mooring, M.; Bosworth, C.M.; Ewing, A.D.; Mantalas, G.L.; Novak, A.M.; van den Bout, A.; Bishara, A.; Rosenkrantz, J.L.; et al. Human-specific NOTCH2NL genes affect notch signaling and cortical neurogenesis. Cell 2018, 173, 1356–1369.e1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, I.K.; Gacquer, D.; Van Heurck, R.; Kumar, D.; Wojno, M.; Bilheu, A.; Herpoel, A.; Lambert, N.; Cheron, J.; Polleux, F.; et al. Human-specific NOTCH2NL genes expand cortical neurogenesis through delta/notch regulation. Cell 2018, 173, 1370–1384.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonacci, F.; Dennis, M.Y.; Huddleston, J.; Sudmant, P.H.; Steinberg, K.M.; Rosenfeld, J.A.; Miroballo, M.; Graves, T.A.; Vives, L.; Malig, M.; et al. Palindromic GOLGA8 core duplicons promote chromosome 15q13.3 microdeletion and evolu-tionary instability. Nat. Genet. 2014, 46, 1293–1302. [Google Scholar] [CrossRef] [Green Version]
- Florio, M.; Albert, M.; Taverna, E.; Namba, T.; Brandl, H.; Lewitus, E.; Haffner, C.; Sykes, A.; Wong, F.K.; Peters, J.; et al. Human-specific gene ARHGAP11B promotes basal progenitor amplification and neocortex expansion. Science 2015, 347, 1465–1470. [Google Scholar] [CrossRef]
- Florio, M.; Namba, T.; Paabo, S.; Hiller, M.; Huttner, W.B. A single splice site mutation in human-specific ARHGAP11B causes basal progenitor amplification. Sci. Adv. 2016, 2, e1601941. [Google Scholar] [CrossRef] [Green Version]
- Heide, M.; Haffner, C.; Murayama, A.; Kurotaki, Y.; Shinohara, H.; Okano, H.; Sasaki, E.; Huttner, W.B. Human-specific ARHGAP11B increases size and folding of primate neocortex in the fetal marmoset. Science 2020, 369, 546–550. [Google Scholar] [CrossRef]
- Namba, T.; Doczi, J.; Pinson, A.; Xing, L.; Kalebic, N.; Wilsch-Brauninger, M.; Long, K.R.; Vaid, S.; Lauer, J.; Bogdanova, A.; et al. Human-specific ARHGAP11B acts in mitochondria to expand neocortical progenitors by glutaminolysis. Neuron 2020, 105, 867–881.e869. [Google Scholar] [CrossRef]
- Raes, J.; Van de Peer, Y. Functional divergence of proteins through frameshift mutations. Trends Genet. 2005, 21, 428–431. [Google Scholar] [CrossRef]
- Xu, G.; Ma, H.; Nei, M.; Kong, H. Evolution of F-box genes in plants: Different modes of sequence divergence and their relationships with functional diversification. Proc. Natl. Acad. Sci. USA 2009, 106, 835–840. [Google Scholar] [CrossRef] [Green Version]
- Guo, B.; Zou, M.; Wagner, A. Pervasive indels and their evolutionary dynamics after the fish-specific genome duplication. Mol. Biol. Evol. 2012, 29, 3005–3022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.; Guo, C.; Shan, H.; Kong, H. Divergence of duplicate genes in exon-intron structure. Proc. Natl. Acad. Sci. USA 2012, 109, 1187–1192. [Google Scholar] [CrossRef] [Green Version]
- Vandenbussche, M.; Theissen, G.; Van de Peer, Y.; Gerats, T. Structural diversification and neo-functionalization during floral MADS-box gene evolution by C-terminal frameshift mutations. Nucleic Acids Res. 2003, 31, 4401–4409. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birney, E.; Clamp, M.; Durbin, R. Genewise and genomewise. Genome Res. 2004, 14, 988–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experi-ments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Teshima, K.M.; Innan, H. The effect of gene conversion on the divergence between duplicated genes. Genetics 2004, 166, 1553–1560. [Google Scholar] [CrossRef] [Green Version]
- Royall, A.H.; Maeso, I.; Dunwell, T.L.; Holland, P.W.H. Mouse Obox and Crxos modulate preimplantation transcriptional profiles revealing similarity between paralogous mouse and human homeobox genes. EvoDevo 2018, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Del Punta, K.; Rothman, A.; Rodriguez, I.; Mombaerts, P. Sequence diversity and genomic organization of vomeronasal receptor genes in the mouse. Genome Res. 2000, 10, 1958–1967. [Google Scholar] [CrossRef]
- Holterhoff, C.K.; Saunders, R.H.; Brito, E.E.; Wagner, D.S. Sequence and expression of the zebrafish α-actinin gene gamily reveals conservation and diversification among Vvertebrates. Dev. Dyn. 2009, 238, 2936–2947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).