
MDSN: A Module Detection Method for Identifying High-Order Epistatic Interactions



Abstract
1. Introduction
2. Materials and Methods
2.1. Epistatic Interaction
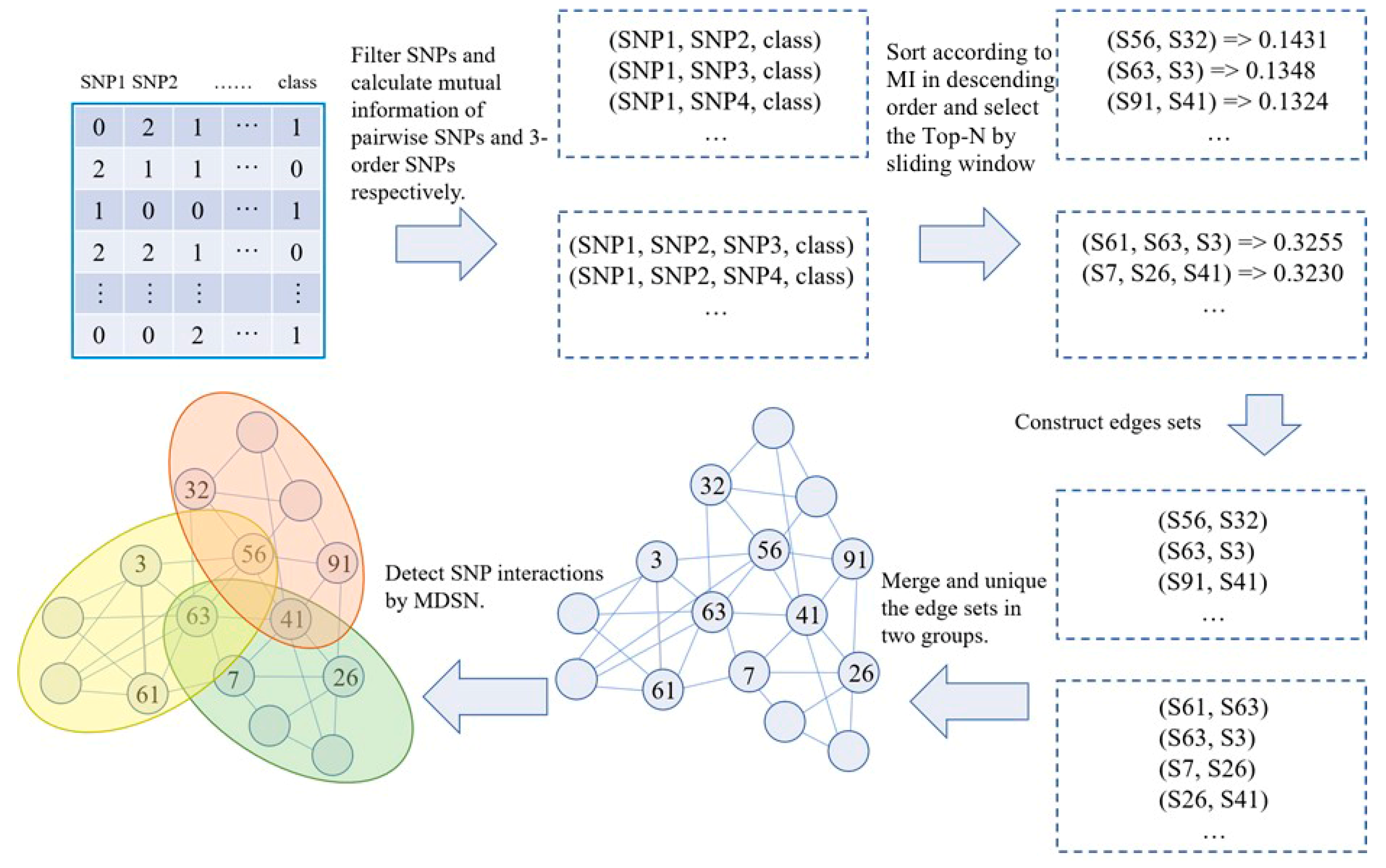
2.2. SNP Network Construction
2.3. Module Detection
3. Results
3.1. Evaluation Criteria
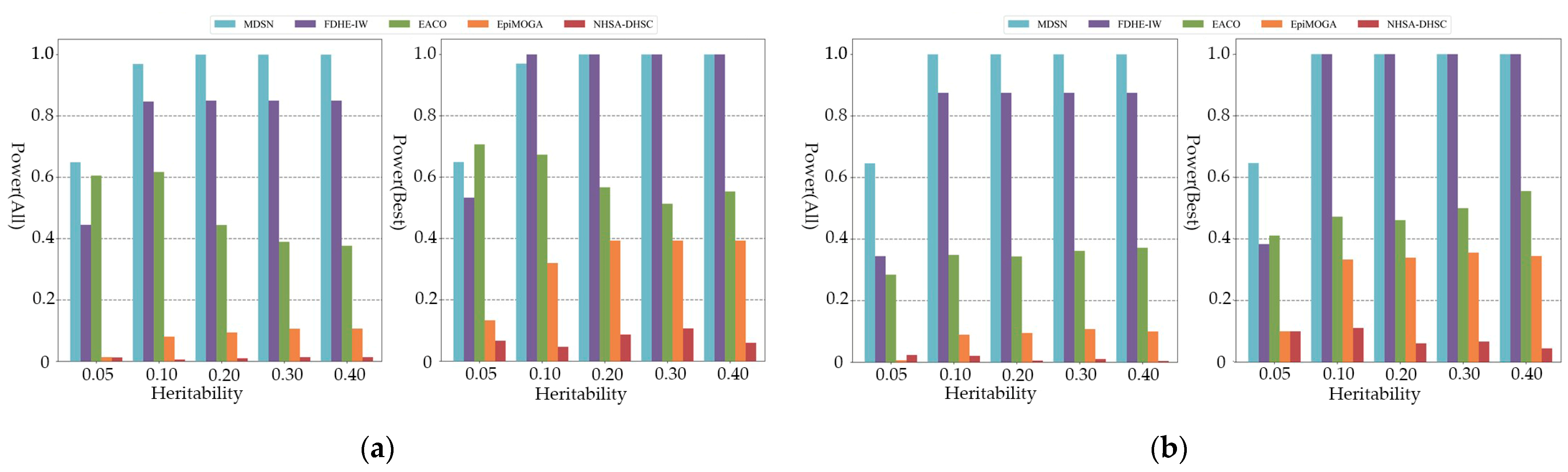
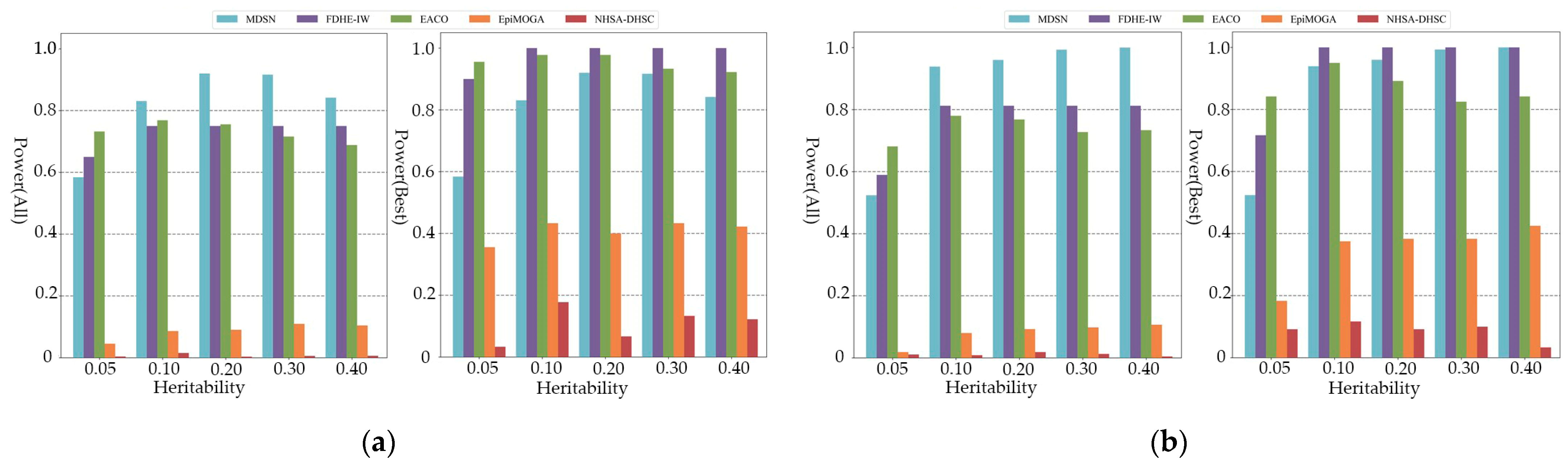
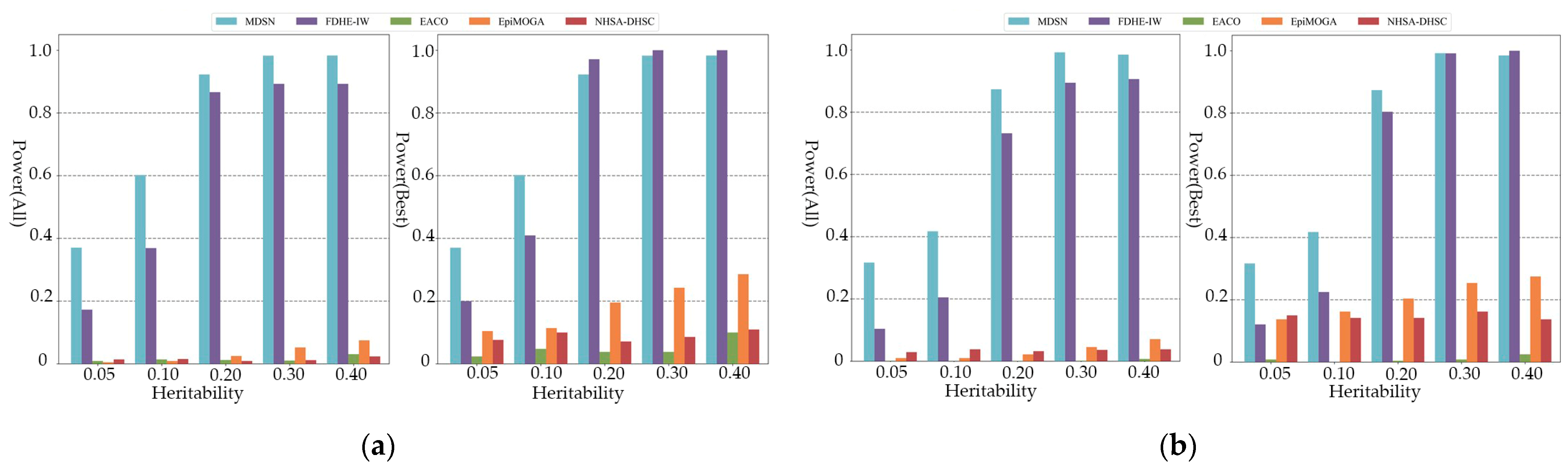
3.2. Experimental on Simulated Data
3.3. Experimental on Real AMD Data

{kind=link}
{kind=link}
{kind=link}
{kind=link}
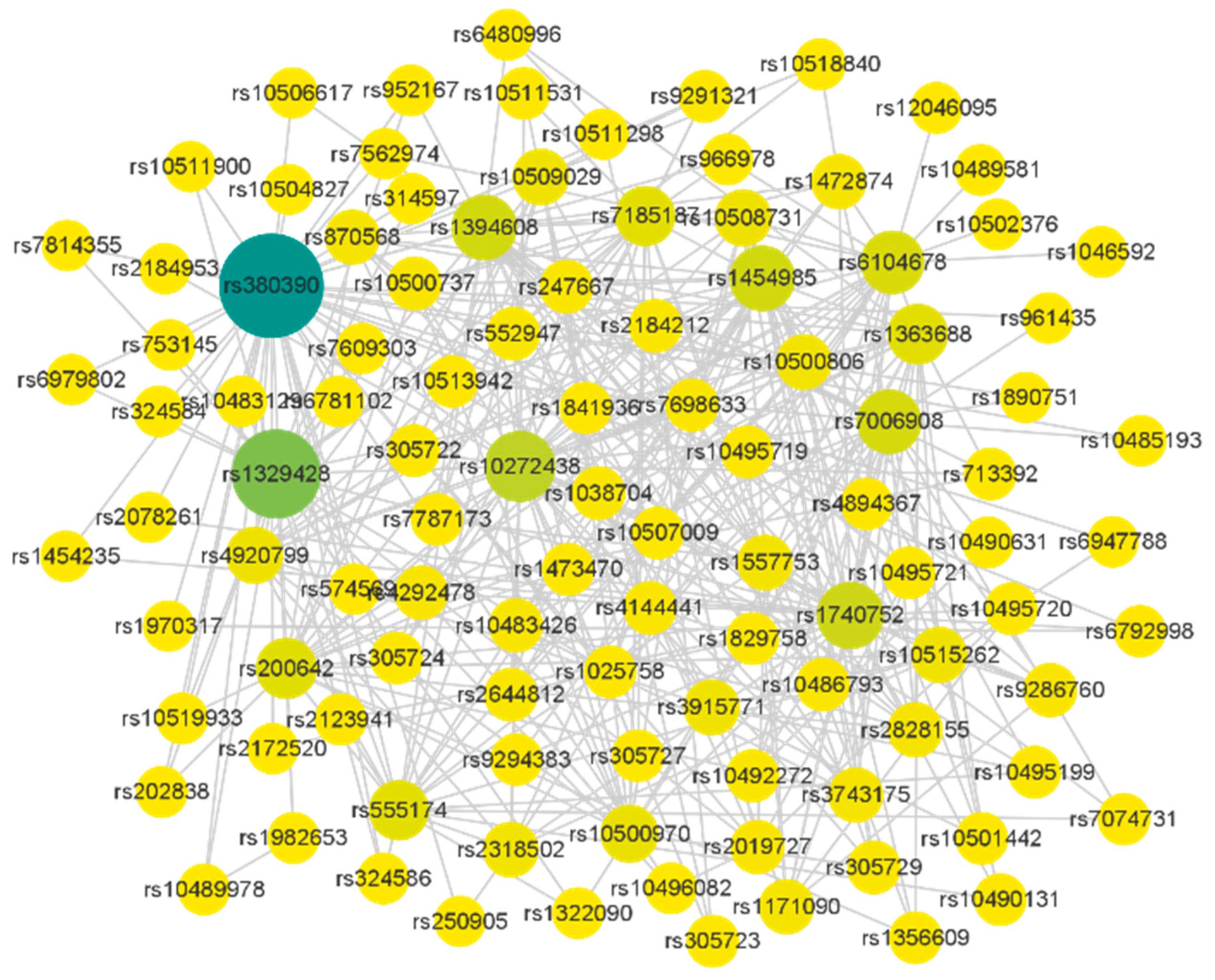{kind=link}
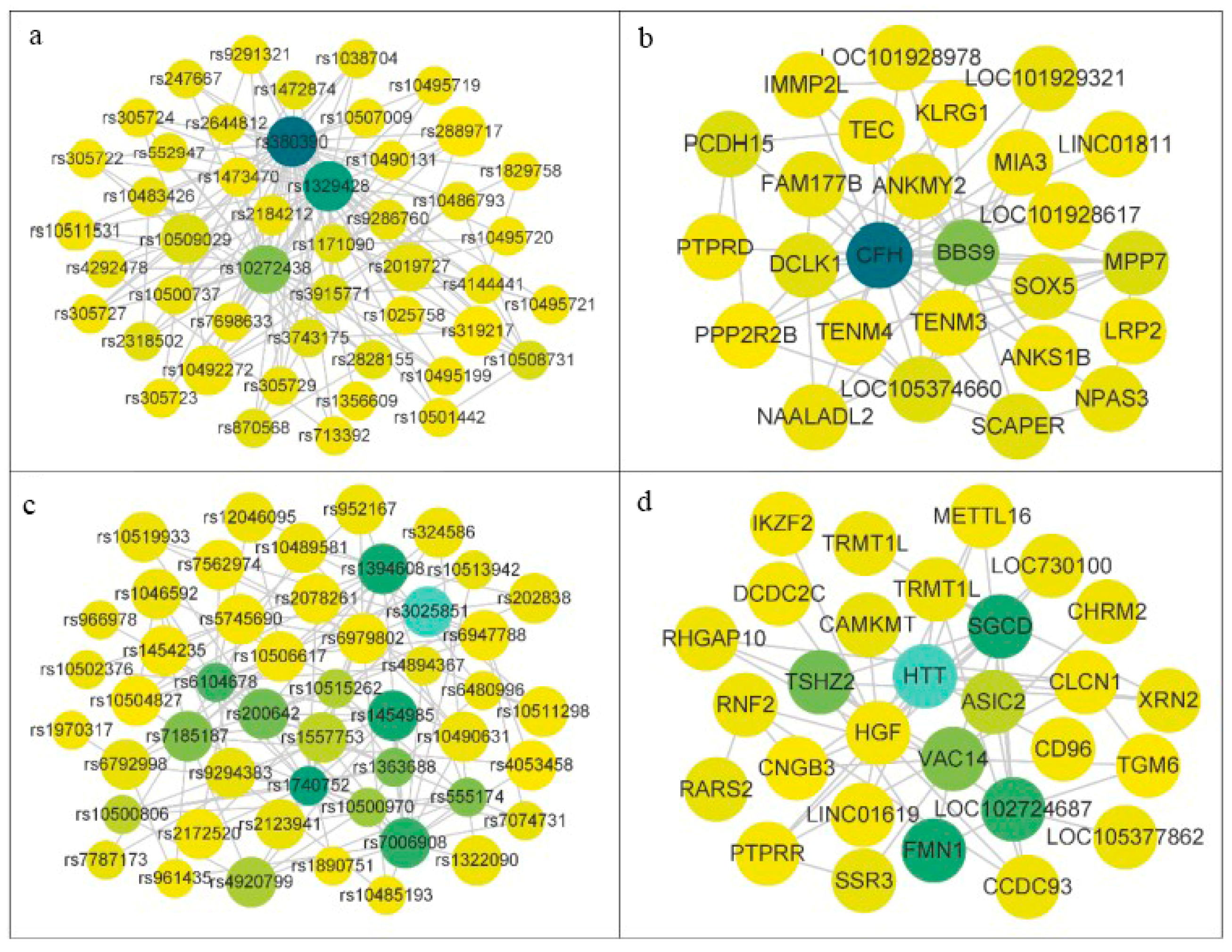{kind=link}
| Module | SNPs | Gene |
|---|---|---|
| 1 | rs6114139, rs1046592, rs2250886, rs1683 147, rs7609303, rs305723, rs1329428, rs12046095 | RNF2, DNHD1, CFH, TRMT1L |
| 2 | rs1046592, rs2250886, rs1683147, rs7609303, rs305723, rs1329428, rs1924257, rs12046095 | RNF2, DNHD1, CFH, TRMTIL, LOC107985255 |
| 3 | rs1046592, rs2250886, rs1683147, rs7609303, rs305723, rs1329428, rs319217, rs6467309, rs2019727, rs12046095 | RNF2, DNHD1, CFH, TRMT1L, PPP2R2B, COPG2 |
| 4 | rs6114139, rs1046592, rs2250886, rs1683147, rs7609303, rs305723, rs1329428, rs10504827, rs6467309, rs2019727, rs12046095 | RNF2, DNHD1, CFH, TRMT1L, CNGB3, COPG2 |
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moore, J.H.; Asselbergs, F.W.; Williams, S.M. Bioinformatics challenges for genome-wide association studies. Bioinformatics 2010, 26, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Wang, J.; Zelikovsky, A.; Guo, X.; Xie, M.; Pan, Y. Searching high-order snp combinations for complex diseases based on energy distribution difference. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 12, 695–704. [Google Scholar] [CrossRef]
- De, R.; Bush, W.S.; Moore, J.H. Bioinformatics challenges in genome-wide association studies (gwas). Clin. Bioinform. 2014, 1168, 63–81. [Google Scholar]
- Ritchie, M.D.; Hahn, L.W.; Roodi, N.; Bailey, L.R.; Dupont, W.D.; Parl, F.F.; Moore, J.H. Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer. Am. J. Hum. Genet. 2001, 69, 138–147. [Google Scholar] [CrossRef]
- Ponte-Fernández, C.; González-Domínguez, J.; Martín, M.J. Fast search of third-order epistatic interactions on cpu and gpu clusters. Int. J. High Perform. Comput. Appl. 2020, 34, 20–29. [Google Scholar] [CrossRef]
- Wan, X.; Yang, C.; Yang, Q.; Xue, H.; Fan, X.; Tang, N.L.; Yu, W. Boost: A fast approach to detecting gene-gene interactions in genome-wide case-control studies. Am. J. Hum. Genet. 2010, 87, 325–340. [Google Scholar] [CrossRef]
- Shang, J.; Zhang, J.; Sun, Y.; Zhang, Y. Epiminer: A three-stage co-information based method for detecting and visualizing epistatic interactions. Digit. Signal Process. 2014, 24, 1–13. [Google Scholar] [CrossRef]
- Liu, J.; Yu, G.; Jiang, Y.; Wang, J. Hiseeker: Detecting high-order snp interactions based on pairwise snp combinations. Genes 2017, 8, 153. [Google Scholar] [CrossRef]
- Guo, X.; Meng, Y.; Yu, N.; Pan, Y. Cloud computing for detecting high-order genome-wide epistatic interaction via dynamic clustering. BMC Bioinform. 2014, 15, 1–16. [Google Scholar] [CrossRef]
- Jing, P.-J.; Shen, H.-B. Macoed: A multi-objective ant colony optimization algorithm for snp epistasis detection in genome-wide association studies. Bioinformatics 2015, 31, 634–641. [Google Scholar] [CrossRef]
- Tuo, S.; Zhang, J.; Yuan, X.; He, Z.; Liu, Y.; Liu, Z. Niche harmony search algorithm for detecting complex disease associated high-order snp combinations. Sci. Rep. 2017, 7, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Tuo, S.; Liu, H.; Chen, H. Multipopulation harmony search algorithm for the detection of high-order snp interactions. Bioinformatics 2020, 36, 4389–4398. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, F.; Pian, C.; Xu, M.; Kong, L.; Fang, J.; Li, Z.; Zhang, L. Epimoga: An epistasis detection method based on a multi-objective genetic algorithm. Genes 2021, 12, 191. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Shang, J.; Liu, J.; Li, S. In An improved ant colony optimization algorithm for the detection of snp-snp interactions. In Proceedings of the International Conference on Intelligent Computing, Lanzhou, China, 2–5 August 2016; Springer: Berlin/Heidelberg, Germany, 2016; pp. 21–32. [Google Scholar]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Liu, G.; Wang, H.; Tian, Y.; Zhou, Z.; Han, L.; Yan, L. Research on single nucleotide polymorphisms interaction detection from network perspective. PLoS ONE 2015, 10, e0119146. [Google Scholar] [CrossRef]
- Wang, J.; Liang, J.; Zheng, W.; Zhao, X.; Mu, J. Protein complex detection algorithm based on multiple topological characteristics in ppi networks. Inf. Sci. 2019, 489, 78–92. [Google Scholar] [CrossRef]
- Yip, D.K.-S.; Chan, L.L.; Pang, I.K.; Jiang, W.; Tang, N.L.; Yu, W.; Yip, K.Y. A network approach to exploring the functional basis of gene–gene epistatic interactions in disease susceptibility. Bioinformatics 2018, 34, 1741–1749. [Google Scholar] [CrossRef]
- Moore, J.H.; Williams, S.M. Epistasis and its implications for personal genetics. Am. J. Hum. Genet. 2009, 85, 309–320. [Google Scholar] [CrossRef]
- Lee, K.-Y.; Leung, K.-S.; Ma, S.L.; So, H.C.; Huang, D.; Tang, N.L.-S.; Wong, M.-H. Genome-wide search for snp interactions in gwas data: Algorithm, feasibility, replication using schizophrenia datasets. Front. Genet. 2020, 11, 1003. [Google Scholar] [CrossRef]
- Granizo-Mackenzie, D.; Moore, J.H. In Multiple threshold spatially uniform relieff for the genetic analysis of complex human diseases. In Proceedings of the European Conference on Evolutionary Computation, Machine Learning and Data Mining in Bioinformatics, Vienna, Austria, 3–5 April 2013; Springer: Berlin/Heidelberg, Germany, 2013; pp. 1–10. [Google Scholar]
- Urbanowicz, R.J.; Kiralis, J.; Sinnott-Armstrong, N.A.; Heberling, T.; Fisher, J.M.; Moore, J.H. Gametes: A fast, direct algorithm for generating pure, strict, epistatic models with random architectures. BioData Min. 2012, 5, 1–14. [Google Scholar] [CrossRef]
- Ponte-Fernández, C.; González-Domínguez, J.; Carvajal-Rodríguez, A.; Martín, M.J. Toxo: A library for calculating penetrance tables of high-order epistasis models. BMC Bioinform. 2020, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tuo, S. Fdhe-iw: A fast approach for detecting high-order epistasis in genome-wide case-control studies. Genes 2018, 9, 435. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, X.; Shang, J.; Liu, J.X.; Zheng, C.H.; Lei, X. Introducing heuristic information into ant colony optimization algorithm for identifying epistasis. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 17, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.-Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.M.; Mayne, S.T. Complement factor h polymorphism in age-related macular degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Wu, X.; Jiang, R.; Li, Y. Epistatic module detection for case-control studies: A bayesian model with a gibbs sampling strategy. PLoS Genet. 2009, 5, e1000464. [Google Scholar] [CrossRef]
- Lin, W.-Y.; Lee, W.-C. Incorporating prior knowledge to facilitate discoveries in a genome-wide association study on age-related macular degeneration. BMC Res. Notes 2010, 3, 1–5. [Google Scholar] [CrossRef]
- Ansari, M.; Mckeigue, P.M.; Skerka, C.; Hayward, C.; Rudan, I.; Vitart, V.; Polasek, O.; Armbrecht, A.-M.; Yates, J.R.W.; Vatavuk, Z.; et al. Genetic influences on plasma cfh and cfhr1 concentrations and their role in susceptibility to age-related macular degeneration. Hum. Mol. Genet. 2013, 22, 4857–4869. [Google Scholar] [CrossRef]
- Rappaport, N.; Twik, M.; Plaschkes, I.; Nudel, R.; Iny Stein, T.; Levitt, J.; Gershoni, M.; Morrey, C.P.; Safran, M.; Lancet, D. Malacards: An amalgamated human disease compendium with diverse clinical and genetic annotation and structured search. Nucleic Acids Res. 2017, 45, D877–D887. [Google Scholar] [CrossRef]
- Matilla-Dueñas, A.; Ashizawa, T.; Brice, A.; Magri, S.; McFarland, K.N.; Pandolfo, M.; Pulst, S.M.; Riess, O.; Rubinsztein, D.C.; Schmidt, J.; et al. Consensus paper: Pathological mechanisms underlying neurodegeneration in spinocerebellar ataxias. Cerebellum 2014, 13, 269–302. [Google Scholar] [CrossRef]
- Birtel, J.; Eisenberger, T.; Gliem, M.; Müller, P.L.; Herrmann, P.; Betz, C.; Zahnleiter, D.; Neuhaus, C.; Lenzner, S.; Holz, F.G.; et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep. 2018, 8, 4824. [Google Scholar] [CrossRef]
- Ong, T.; Pennesi, M.; Birch, D.; Lam, B.; Tsang, S. Adeno-associated viral gene therapy for inherited retinal disease. Pharm. Res. 2019, 36, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Luo, L.; Xing, C.; Chen, Y.; Xu, P.; Li, M.; Zeng, L.; Li, C.; Ghosh, S.; Della Manna, D. Rnf2 ablation reprograms the tumor-immune microenvironment and stimulates durable nk and cd4+ t-cell-dependent antitumor immunity. Nat. Cancer 2021, 2, 1018–1038. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Meng, L.; Lv, M.; He, X.; Sha, Y.; Tang, D.; Tan, Y.; Hu, T.; He, W.; Tu, C. Bi-allelic variants in dnhd1 cause flagellar axoneme defects and asthenoteratozoospermia in humans and mice. Am. J. Hum. Genet. 2022, 109, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Jonkhout, N.; Cruciani, S.; Santos Vieira, H.G.; Tran, J.; Liu, H.; Liu, G.; Pickford, R.; Kaczorowski, D.; Franco, G.R.; Vauti, F. Subcellular relocalization and nuclear redistribution of the rna methyltransferases trmt1 and trmt1l upon neuronal activation. RNA Biol. 2021, 18, 1905–1919. [Google Scholar] [CrossRef] [PubMed]






| Model | MDSN | FDHE-IW | EACO | EpiMOGA | NHSA-DHSC |
|---|---|---|---|---|---|
| Additive-5 | 36.64 | 298.46 | 246.19 | 416.47 | 66.01 |
| Additive-6 | 37.60 | 691.08 | 241.08 | 454.04 | 66.28 |
| Multiplicative-3 | 37.47 | 49.88 | 241.45 | 296.80 | 76.75 |
| Multiplicative-4 | 36.96 | 124.20 | 241.36 | 374.74 | 68.89 |
| Threshold-7 | 39.02 | 1375.88 | 241.47 | 487.88 | 66.50 |
| Threshold-8 | 38.94 | 2875.88 | 241.59 | 513.62 | 92.53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Gu, Y.; Ren, Q.; Li, Y.; Shang, J.; Liu, J.-X.; Guan, B. MDSN: A Module Detection Method for Identifying High-Order Epistatic Interactions. Genes 2022, 13, 2403. https://doi.org/10.3390/genes13122403
Sun Y, Gu Y, Ren Q, Li Y, Shang J, Liu J-X, Guan B. MDSN: A Module Detection Method for Identifying High-Order Epistatic Interactions. Genes. 2022; 13(12):2403. https://doi.org/10.3390/genes13122403
Chicago/Turabian StyleSun, Yan, Yijun Gu, Qianqian Ren, Yiting Li, Junliang Shang, Jin-Xing Liu, and Boxin Guan. 2022. "MDSN: A Module Detection Method for Identifying High-Order Epistatic Interactions" Genes 13, no. 12: 2403. https://doi.org/10.3390/genes13122403
APA StyleSun, Y., Gu, Y., Ren, Q., Li, Y., Shang, J., Liu, J.-X., & Guan, B. (2022). MDSN: A Module Detection Method for Identifying High-Order Epistatic Interactions. Genes, 13(12), 2403. https://doi.org/10.3390/genes13122403