

Fatty Acid Metabolism in Endothelial Cell


Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Role of Endothelial Cells in Angiogenesis, Homeostasis, Stimulus/Immune Response
3. Endothelial Cell Metabolism
3.1. Endothelial Cell Glucose and Amino Acid Metabolism
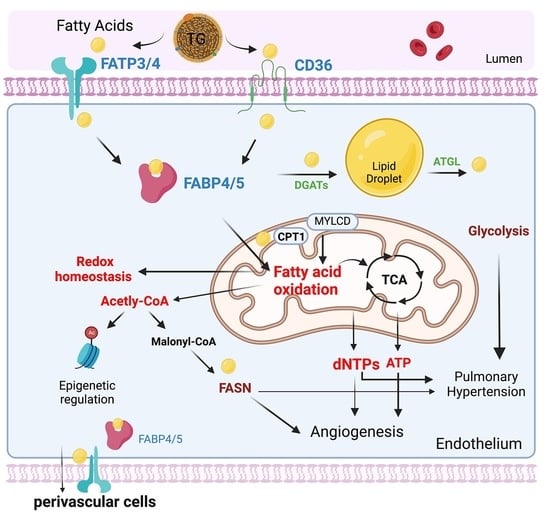
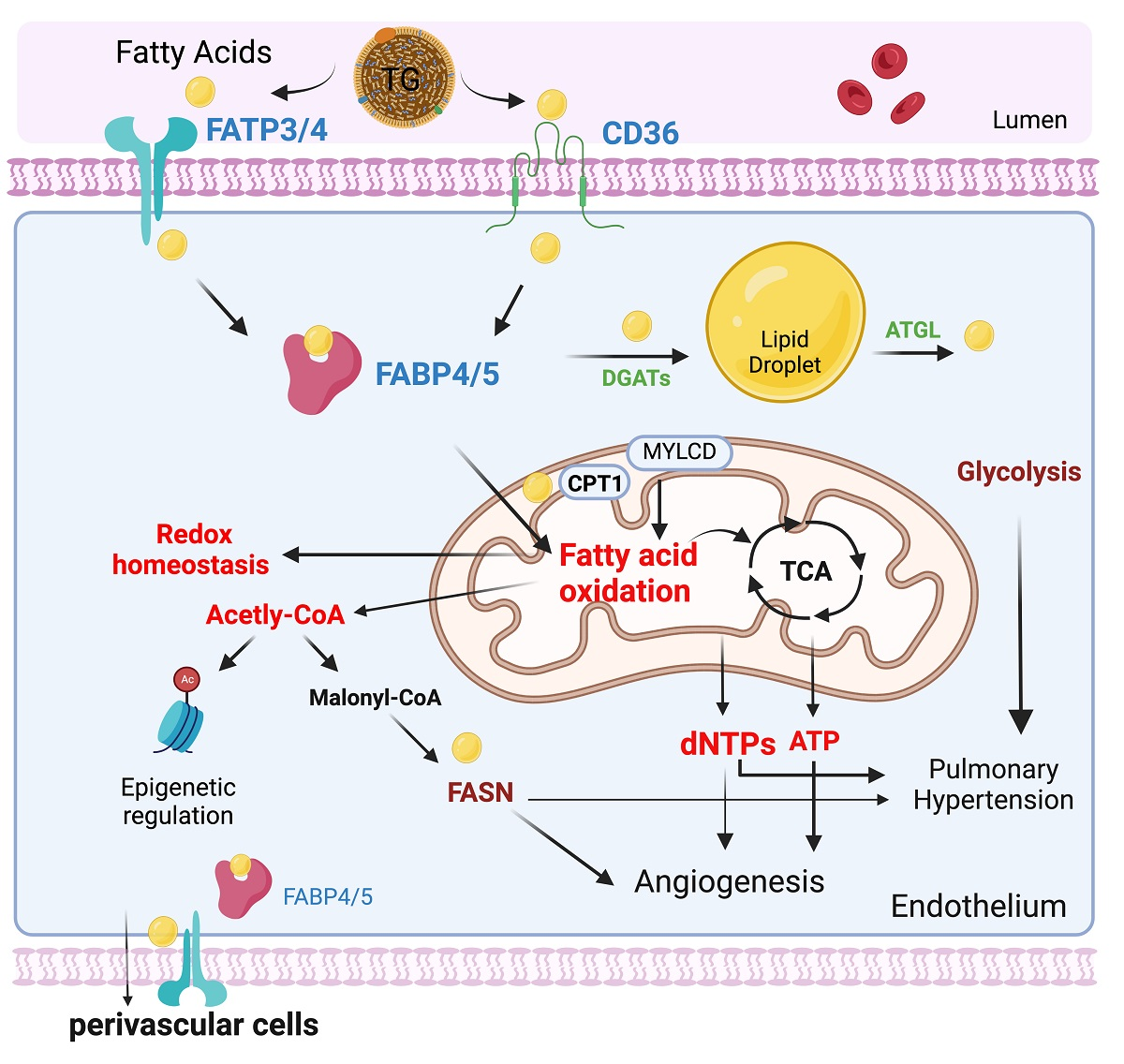
3.2. Endothelial Cell Fatty Acids (FAs) Metabolism
3.2.1. Endothelial Cell Fatty Acid Uptake and Transport
CD36
FATPs
The FABPs
3.2.2. Endothelial Fatty Acid Storage and Lipolysis
3.2.3. Endothelial Cell Fatty Acid Oxidation
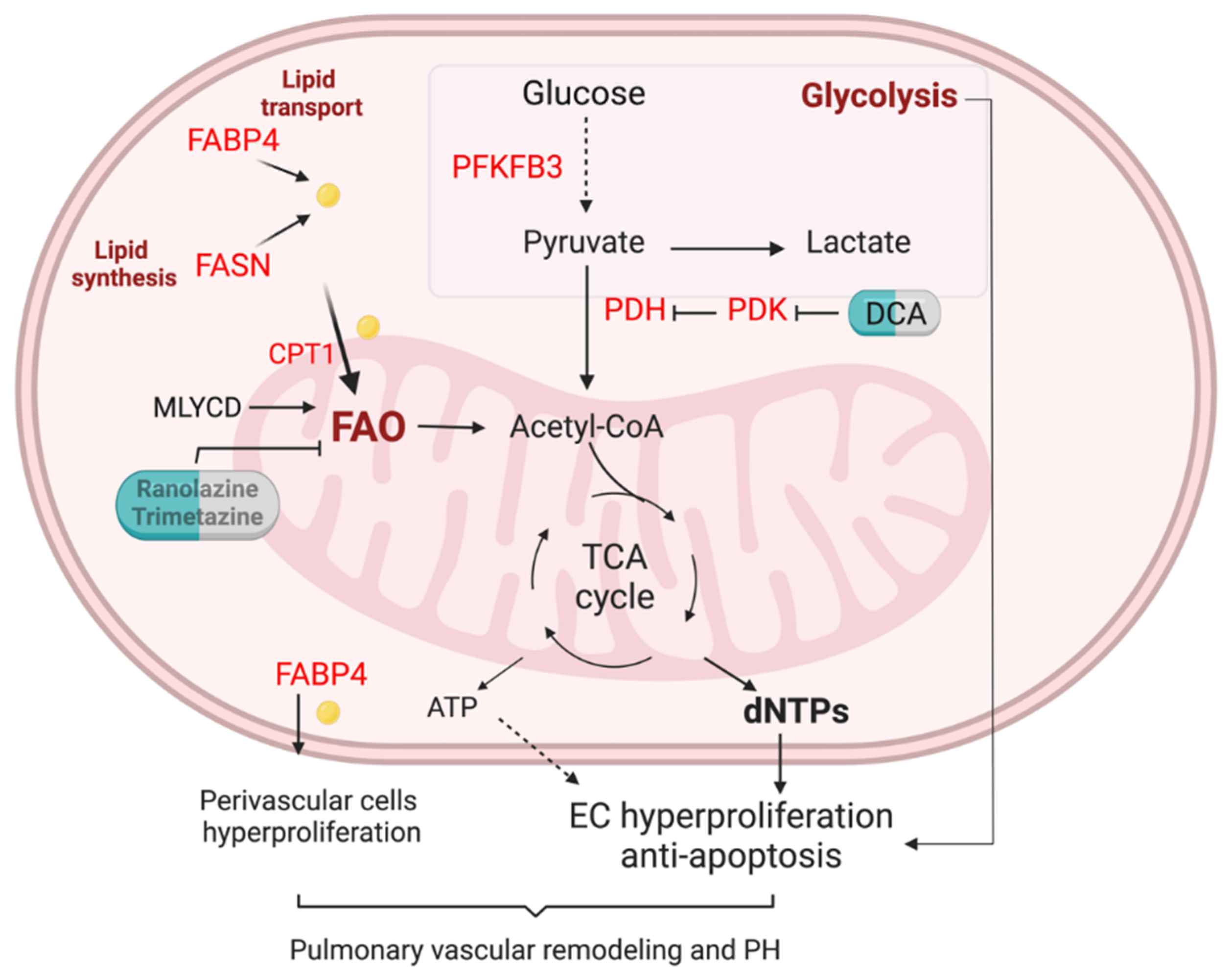
3.3. Fatty Acid Metabolism in Pulmonary Hypertension
4. Therapeutic Opportunities of FA Metabolism in PH
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rohlenova, K.; Veys, K.; Miranda-Santos, I.; De Bock, K.; Carmeliet, P. Endothelial Cell Metabolism in Health and Disease. Trends Cell Biol. 2018, 28, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Eelen, G.; de Zeeuw, P.; Simons, M.; Carmeliet, P. Endothelial cell metabolism in normal and diseased vasculature. Circ. Res. 2015, 116, 1231–1244. [Google Scholar] [CrossRef] [PubMed]
- Pircher, A.; Treps, L.; Bodrug, N.; Carmeliet, P. Endothelial cell metabolism: A novel player in atherosclerosis? Basic principles and therapeutic opportunities. Atherosclerosis 2016, 253, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, X.; Carmeliet, P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metab. 2019, 30, 414–433. [Google Scholar] [CrossRef]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef]
- Eelen, G.; Treps, L.; Li, X.; Carmeliet, P. Basic and Therapeutic Aspects of Angiogenesis Updated. Circ. Res. 2020, 127, 310–329. [Google Scholar] [CrossRef]
- Rafii, S.; Butler, J.M.; Ding, B.S. Angiocrine functions of organ-specific endothelial cells. Nature 2016, 529, 316–325. [Google Scholar] [CrossRef]
- Jakobsson, L.; Franco, C.A.; Bentley, K.; Collins, R.T.; Ponsioen, B.; Aspalter, I.M.; Rosewell, I.; Busse, M.; Thurston, G.; Medvinsky, A.; et al. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat. Cell Biol. 2010, 12, 943–953. [Google Scholar] [CrossRef]
- Jaffe, E.A. Cell biology of endothelial cells. Hum. Pathol. 1987, 18, 234–239. [Google Scholar] [CrossRef]
- Steinberg, H.O.; Paradisi, G.; Hook, G.; Crowder, K.; Cronin, J.; Baron, A.D. Free fatty acid elevation impairs insulin-mediated vasodilation and nitric oxide production. Diabetes 2000, 49, 1231–1238. [Google Scholar] [CrossRef]
- Leo, F.; Suvorava, T.; Heuser, S.K.; Li, J.; LoBue, A.; Barbarino, F.; Piragine, E.; Schneckmann, R.; Hutzler, B.; Good, M.E.; et al. Red Blood Cell and Endothelial eNOS Independently Regulate Circulating Nitric Oxide Metabolites and Blood Pressure. Circulation 2021, 144, 870–889. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837, 837a–837d. [Google Scholar] [CrossRef] [PubMed]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Opitz, B.; Förster, S.; Hocke, A.C.; Maass, M.; Schmeck, B.; Hippenstiel, S.; Suttorp, N.; Krüll, M. Nod1-mediated endothelial cell activation by Chlamydophila pneumoniae. Circ. Res. 2005, 96, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kumar, A.; Carmeliet, P. Metabolic Pathways Fueling the Endothelial Cell Drive. Annu. Rev. Physiol. 2019, 81, 483–503. [Google Scholar] [CrossRef] [PubMed]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquière, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [PubMed]
- De Bock, K.; Georgiadou, M.; Carmeliet, P. Role of endothelial cell metabolism in vessel sprouting. Cell Metab. 2013, 18, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Helmlinger, G.; Endo, M.; Ferrara, N.; Hlatky, L.; Jain, R.K. Formation of endothelial cell networks. Nature 2000, 405, 139–141. [Google Scholar] [CrossRef]
- Yu, P.; Wilhelm, K.; Dubrac, A.; Tung, J.K.; Alves, T.C.; Fang, J.S.; Xie, Y.; Zhu, J.; Chen, Z.; De Smet, F.; et al. FGF-dependent metabolic control of vascular development. Nature 2017, 545, 224–228. [Google Scholar] [CrossRef]
- Kim, B.; Li, J.; Jang, C.; Arany, Z. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. 2017, 36, 2321–2333. [Google Scholar] [CrossRef]
- Huang, H.; Vandekeere, S.; Kalucka, J.; Bierhansl, L.; Zecchin, A.; Brüning, U.; Visnagri, A.; Yuldasheva, N.; Goveia, J.; Cruys, B.; et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017, 36, 2334–2352. [Google Scholar] [CrossRef] [PubMed]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, C.E.; Falkevall, A.; Wang, X.; Larsson, E.; Huusko, J.; Nilsson, I.; van Meeteren, L.A.; Samen, E.; Lu, L.; Vanwildemeersch, M.; et al. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature 2010, 464, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Kazantzis, M.; Stahl, A. Fatty acid transport proteins, implications in physiology and disease. Biochim. Biophys. Acta 2012, 1821, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Hülsmann, W.C.; Dubelaar, M.L. Aspects of fatty acid metabolism in vascular endothelial cells. Biochimie 1988, 70, 681–686. [Google Scholar] [CrossRef]
- Dallinga-Thie, G.M.; Franssen, R.; Mooij, H.L.; Visser, M.E.; Hassing, H.C.; Peelman, F.; Kastelein, J.J.; Péterfy, M.; Nieuwdorp, M. The metabolism of triglyceride-rich lipoproteins revisited: New players, new insight. Atherosclerosis 2010, 211, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.S.; Beigneux, A.P.; Barnes, R.H., 2nd; Tu, Y.; Gin, P.; Weinstein, M.M.; Nobumori, C.; Nyrén, R.; Goldberg, I.; Olivecrona, G.; et al. GPIHBP1 is responsible for the entry of lipoprotein lipase into capillaries. Cell Metab. 2010, 12, 42–52. [Google Scholar] [CrossRef]
- Martin, C.; Chevrot, M.; Poirier, H.; Passilly-Degrace, P.; Niot, I.; Besnard, P. CD36 as a lipid sensor. Physiol. Behav. 2011, 105, 36–42. [Google Scholar] [CrossRef]
- Hagberg, C.; Mehlem, A.; Falkevall, A.; Muhl, L.; Eriksson, U. Endothelial fatty acid transport: Role of vascular endothelial growth factor B. Physiology 2013, 28, 125–134. [Google Scholar] [CrossRef]
- Smathers, R.L.; Petersen, D.R. The human fatty acid-binding protein family: Evolutionary divergences and functions. Hum. Genom. 2011, 5, 170–191. [Google Scholar] [CrossRef]
- Storch, J.; Corsico, B. The emerging functions and mechanisms of mammalian fatty acid-binding proteins. Annu. Rev. Nutr. 2008, 28, 73–95. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, M.; Hotamisligil, G.S. Fatty acid-binding proteins: Role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discov. 2008, 7, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Canton, J.; Neculai, D.; Grinstein, S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 2013, 13, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, R.L.; Febbraio, M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2009, 2, re3. [Google Scholar] [CrossRef] [PubMed]
- Hoebe, K.; Georgel, P.; Rutschmann, S.; Du, X.; Mudd, S.; Crozat, K.; Sovath, S.; Shamel, L.; Hartung, T.; Zähringer, U.; et al. CD36 is a sensor of diacylglycerides. Nature 2005, 433, 523–527. [Google Scholar] [CrossRef]
- Asch, A.S.; Barnwell, J.; Silverstein, R.L.; Nachman, R.L. Isolation of the thrombospondin membrane receptor. J. Clin. Investig. 1987, 79, 1054–1061. [Google Scholar] [CrossRef]
- Hao, J.W.; Wang, J.; Guo, H.; Zhao, Y.Y.; Sun, H.H.; Li, Y.F.; Lai, X.Y.; Zhao, N.; Wang, X.; Xie, C.; et al. CD36 facilitates fatty acid uptake by dynamic palmitoylation-regulated endocytosis. Nat. Commun. 2020, 11, 4765. [Google Scholar] [CrossRef]
- Bastie, C.C.; Hajri, T.; Drover, V.A.; Grimaldi, P.A.; Abumrad, N.A. CD36 in myocytes channels fatty acids to a lipase-accessible triglyceride pool that is related to cell lipid and insulin responsiveness. Diabetes 2004, 53, 2209–2216. [Google Scholar] [CrossRef]
- Wang, J.; Hao, J.W.; Wang, X.; Guo, H.; Sun, H.H.; Lai, X.Y.; Liu, L.Y.; Zhu, M.; Wang, H.Y.; Li, Y.F.; et al. DHHC4 and DHHC5 Facilitate Fatty Acid Uptake by Palmitoylating and Targeting CD36 to the Plasma Membrane. Cell Rep. 2019, 26, 209–221.e5. [Google Scholar] [CrossRef]
- Goldberg, I.J.; Eckel, R.H.; Abumrad, N.A. Regulation of fatty acid uptake into tissues: Lipoprotein lipase- and CD36-mediated pathways. J. Lipid Res. 2009, 50, S86–S90. [Google Scholar] [CrossRef]
- Son, N.H.; Basu, D.; Samovski, D.; Pietka, T.A.; Peche, V.S.; Willecke, F.; Fang, X.; Yu, S.Q.; Scerbo, D.; Chang, H.R.; et al. Endothelial cell CD36 optimizes tissue fatty acid uptake. J. Clin. Investig. 2018, 128, 4329–4342. [Google Scholar] [CrossRef] [PubMed]
- Bou Khzam, L.; Son, N.H.; Mullick, A.E.; Abumrad, N.A.; Goldberg, I.J. Endothelial cell CD36 deficiency prevents normal angiogenesis and vascular repair. Am. J. Transl. Res. 2020, 12, 7737–7761. [Google Scholar] [PubMed]
- Ramakrishnan, D.P.; Hajj-Ali, R.A.; Chen, Y.; Silverstein, R.L. Extracellular Vesicles Activate a CD36-Dependent Signaling Pathway to Inhibit Microvascular Endothelial Cell Migration and Tube Formation. Arter. Thromb. Vasc. Biol. 2016, 36, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Rekhi, U.R.; Omar, M.; Alexiou, M.; Delyea, C.; Immaraj, L.; Elahi, S.; Febbraio, M. Endothelial Cell CD36 Reduces Atherosclerosis and Controls Systemic Metabolism. Front. Cardiovasc. Med. 2021, 8, 768481. [Google Scholar] [CrossRef]
- Cifarelli, V.; Appak-Baskoy, S.; Peche, V.S.; Kluzak, A.; Shew, T.; Narendran, R.; Pietka, K.M.; Cella, M.; Walls, C.W.; Czepielewski, R.; et al. Visceral obesity and insulin resistance associate with CD36 deletion in lymphatic endothelial cells. Nat. Commun. 2021, 12, 3350. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Oh, S.F.; Wada, S.; Rowe, G.C.; Liu, L.; Chan, M.C.; Rhee, J.; Hoshino, A.; Kim, B.; Ibrahim, A.; et al. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat. Med. 2016, 22, 421–426. [Google Scholar] [CrossRef]
- Bae, H.; Hong, K.Y.; Lee, C.K.; Jang, C.; Lee, S.J.; Choe, K.; Offermanns, S.; He, Y.; Lee, H.J.; Koh, G.Y. Angiopoietin-2-integrin α5β1 signaling enhances vascular fatty acid transport and prevents ectopic lipid-induced insulin resistance. Nat. Commun. 2020, 11, 2980. [Google Scholar] [CrossRef]
- Ibrahim, A.; Yucel, N.; Kim, B.; Arany, Z. Local Mitochondrial ATP Production Regulates Endothelial Fatty Acid Uptake and Transport. Cell Metab. 2020, 32, 309–319.e7. [Google Scholar] [CrossRef]
- Lee, C.H.; Lui, D.T.W.; Lam, K.S.L. Adipocyte Fatty Acid-Binding Protein, Cardiovascular Diseases and Mortality. Front. Immunol. 2021, 12, 589206. [Google Scholar] [CrossRef]
- Elmasri, H.; Karaaslan, C.; Teper, Y.; Ghelfi, E.; Weng, M.; Ince, T.A.; Kozakewich, H.; Bischoff, J.; Cataltepe, S. Fatty acid binding protein 4 is a target of VEGF and a regulator of cell proliferation in endothelial cells. FASEB J. 2009, 23, 3865–3873. [Google Scholar] [CrossRef]
- Masouyé, I.; Hagens, G.; Van Kuppevelt, T.H.; Madsen, P.; Saurat, J.H.; Veerkamp, J.H.; Pepper, M.S.; Siegenthaler, G. Endothelial cells of the human microvasculature express epidermal fatty acid-binding protein. Circ. Res. 1997, 81, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Iso, T.; Maeda, K.; Hanaoka, H.; Suga, T.; Goto, K.; Syamsunarno, M.R.; Hishiki, T.; Nagahata, Y.; Matsui, H.; Arai, M.; et al. Capillary endothelial fatty acid binding proteins 4 and 5 play a critical role in fatty acid uptake in heart and skeletal muscle. Arter. Thromb. Vasc. Biol. 2013, 33, 2549–2557. [Google Scholar] [CrossRef] [PubMed]
- Elmasri, H.; Ghelfi, E.; Yu, C.W.; Traphagen, S.; Cernadas, M.; Cao, H.; Shi, G.P.; Plutzky, J.; Sahin, M.; Hotamisligil, G.; et al. Endothelial cell-fatty acid binding protein 4 promotes angiogenesis: Role of stem cell factor/c-kit pathway. Angiogenesis 2012, 15, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Aragonès, G.; Saavedra, P.; Heras, M.; Cabré, A.; Girona, J.; Masana, L. Fatty acid-binding protein 4 impairs the insulin-dependent nitric oxide pathway in vascular endothelial cells. Cardiovasc. Diabetol. 2012, 11, 72. [Google Scholar] [CrossRef]
- Fuseya, T.; Furuhashi, M.; Matsumoto, M.; Watanabe, Y.; Hoshina, K.; Mita, T.; Ishimura, S.; Tanaka, M.; Miura, T. Ectopic Fatty Acid-Binding Protein 4 Expression in the Vascular Endothelium is Involved in Neointima Formation After Vascular Injury. J. Am. Heart Assoc. 2017, 6, e006377. [Google Scholar] [CrossRef]
- Lee, M.Y.; Li, H.; Xiao, Y.; Zhou, Z.; Xu, A.; Vanhoutte, P.M. Chronic administration of BMS309403 improves endothelial function in apolipoprotein E-deficient mice and in cultured human endothelial cells. Br. J. Pharmacol. 2011, 162, 1564–1576. [Google Scholar] [CrossRef]
- Furuhashi, M.; Tuncman, G.; Görgün, C.Z.; Makowski, L.; Atsumi, G.; Vaillancourt, E.; Kono, K.; Babaev, V.R.; Fazio, S.; Linton, M.F.; et al. Treatment of diabetes and atherosclerosis by inhibiting fatty-acid-binding protein aP2. Nature 2007, 447, 959–965. [Google Scholar] [CrossRef]
- Luis, G.; Godfroid, A.; Nishiumi, S.; Cimino, J.; Blacher, S.; Maquoi, E.; Wery, C.; Collignon, A.; Longuespée, R.; Montero-Ruiz, L.; et al. Tumor resistance to ferroptosis driven by Stearoyl-CoA Desaturase-1 (SCD1) in cancer cells and Fatty Acid Biding Protein-4 (FABP4) in tumor microenvironment promote tumor recurrence. Redox Biol. 2021, 43, 102006. [Google Scholar] [CrossRef]
- Boord, J.B.; Maeda, K.; Makowski, L.; Babaev, V.R.; Fazio, S.; Linton, M.F.; Hotamisligil, G.S. Combined adipocyte-macrophage fatty acid-binding protein deficiency improves metabolism, atherosclerosis, and survival in apolipoprotein E-deficient mice. Circulation 2004, 110, 1492–1498. [Google Scholar] [CrossRef]
- Maeda, K.; Cao, H.; Kono, K.; Gorgun, C.Z.; Furuhashi, M.; Uysal, K.T.; Cao, Q.; Atsumi, G.; Malone, H.; Krishnan, B.; et al. Adipocyte/macrophage fatty acid binding proteins control integrated metabolic responses in obesity and diabetes. Cell Metab. 2005, 1, 107–119. [Google Scholar] [CrossRef]
- Yu, C.W.; Liang, X.; Lipsky, S.; Karaaslan, C.; Kozakewich, H.; Hotamisligil, G.S.; Bischoff, J.; Cataltepe, S. Dual role of fatty acid-binding protein 5 on endothelial cell fate: A potential link between lipid metabolism and angiogenic responses. Angiogenesis 2016, 19, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Short, J.L.; Choy, K.H.; Zeng, A.X.; Marriott, P.J.; Owada, Y.; Scanlon, M.J.; Porter, C.J.; Nicolazzo, J.A. Fatty Acid-Binding Protein 5 at the Blood-Brain Barrier Regulates Endogenous Brain Docosahexaenoic Acid Levels and Cognitive Function. J. Neurosci. 2016, 36, 11755–11767. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Iso, T.; Hanaoka, H.; Yamaguchi, A.; Suga, T.; Hattori, A.; Irie, Y.; Shinagawa, Y.; Matsui, H.; Syamsunarno, M.R.; et al. Peroxisome proliferator-activated receptor-γ in capillary endothelia promotes fatty acid uptake by heart during long-term fasting. J. Am. Heart Assoc. 2013, 2, e004861. [Google Scholar] [CrossRef] [PubMed]
- Jabs, M.; Rose, A.J.; Lehmann, L.H.; Taylor, J.; Moll, I.; Sijmonsma, T.P.; Herberich, S.E.; Sauer, S.W.; Poschet, G.; Federico, G.; et al. Inhibition of Endothelial Notch Signaling Impairs Fatty Acid Transport and Leads to Metabolic and Vascular Remodeling of the Adult Heart. Circulation 2018, 137, 2592–2608. [Google Scholar] [CrossRef] [PubMed]
- Hwangbo, C.; Wu, J.; Papangeli, I.; Adachi, T.; Sharma, B.; Park, S.; Zhao, L.; Ju, H.; Go, G.W.; Cui, G.; et al. Endothelial APLNR regulates tissue fatty acid uptake and is essential for apelin’s glucose-lowering effects. Sci. Transl. Med. 2017, 9, eaad4000. [Google Scholar] [CrossRef] [PubMed]
- Jarc, E.; Petan, T. Lipid Droplets and the Management of Cellular Stress. Yale J. Biol. Med. 2019, 92, 435–452. [Google Scholar] [PubMed]
- Kuo, A.; Lee, M.Y.; Sessa, W.C. Lipid Droplet Biogenesis and Function in the Endothelium. Circ. Res. 2017, 120, 1289–1297. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol. 2015, 17, 759–770. [Google Scholar] [CrossRef]
- Schrammel, A.; Mussbacher, M.; Wölkart, G.; Stessel, H.; Pail, K.; Winkler, S.; Schweiger, M.; Haemmerle, G.; Al Zoughbi, W.; Höfler, G.; et al. Endothelial dysfunction in adipose triglyceride lipase deficiency. Biochim. Biophys. Acta 2014, 1841, 906–917. [Google Scholar] [CrossRef]
- Riederer, M.; Lechleitner, M.; Köfeler, H.; Frank, S. Reduced expression of adipose triglyceride lipase decreases arachidonic acid release and prostacyclin secretion in human aortic endothelial cells. Arch. Physiol. Biochem. 2017, 123, 249–253. [Google Scholar] [CrossRef]
- Gogg, S.; Nerstedt, A.; Boren, J.; Smith, U. Human adipose tissue microvascular endothelial cells secrete PPARγ ligands and regulate adipose tissue lipid uptake. JCI Insight 2019, 4, e125914. [Google Scholar] [CrossRef]
- Dagher, Z.; Ruderman, N.; Tornheim, K.; Ido, Y. Acute regulation of fatty acid oxidation and amp-activated protein kinase in human umbilical vein endothelial cells. Circ. Res. 2001, 88, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Kalucka, J.; Bierhansl, L.; Conchinha, N.V.; Missiaen, R.; Elia, I.; Brüning, U.; Scheinok, S.; Treps, L.; Cantelmo, A.R.; Dubois, C.; et al. Quiescent Endothelial Cells Upregulate Fatty Acid β-Oxidation for Vasculoprotection via Redox Homeostasis. Cell Metab. 2018, 28, 881–894.e13. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Gong, J.; Peterson, A.L.; Lu, X.; Zhang, P.; Dennery, P.A. Fatty Acid Oxidation Protects against Hyperoxia-induced Endothelial Cell Apoptosis and Lung Injury in Neonatal Mice. Am. J. Respir. Cell Mol. Biol. 2019, 60, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Edelstein, D.; Obici, S.; Higham, N.; Zou, M.H.; Brownlee, M. Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation. J. Clin. Investig. 2006, 116, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.W.; Wang, X.; Zecchin, A.; Thienpont, B.; Cornelissen, I.; Kalucka, J.; García-Caballero, M.; Missiaen, R.; Huang, H.; Brüning, U.; et al. The role of fatty acid β-oxidation in lymphangiogenesis. Nature 2017, 542, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Zecchin, A.; Wong, B.W.; Tembuyser, B.; Souffreau, J.; Van Nuffelen, A.; Wyns, S.; Vinckier, S.; Carmeliet, P.; Dewerchin, M. Live imaging reveals a conserved role of fatty acid β-oxidation in early lymphatic development in zebrafish. Biochem. Biophys. Res. Commun. 2018, 503, 26–31. [Google Scholar] [CrossRef]
- Xiong, J.; Kawagishi, H.; Yan, Y.; Liu, J.; Wells, Q.S.; Edmunds, L.R.; Fergusson, M.M.; Yu, Z.X.; Rovira, I.I.; Brittain, E.L.; et al. A Metabolic Basis for Endothelial-to-Mesenchymal Transition. Mol. Cell 2018, 69, 689–698.e7. [Google Scholar] [CrossRef]
- Singh, N.; Singh, H.; Jagavelu, K.; Wahajuddin, M.; Hanif, K. Fatty acid synthase modulates proliferation, metabolic functions and angiogenesis in hypoxic pulmonary artery endothelial cells. Eur. J. Pharmacol. 2017, 815, 462–469. [Google Scholar] [CrossRef]
- Bruning, U.; Morales-Rodriguez, F.; Kalucka, J.; Goveia, J.; Taverna, F.; Queiroz, K.C.S.; Dubois, C.; Cantelmo, A.R.; Chen, R.; Loroch, S.; et al. Impairment of Angiogenesis by Fatty Acid Synthase Inhibition Involves mTOR Malonylation. Cell Metab. 2018, 28, 866–880.e15. [Google Scholar] [CrossRef]
- Wei, X.; Schneider, J.G.; Shenouda, S.M.; Lee, A.; Towler, D.A.; Chakravarthy, M.V.; Vita, J.A.; Semenkovich, C.F. De novo lipogenesis maintains vascular homeostasis through endothelial nitric-oxide synthase (eNOS) palmitoylation. J. Biol. Chem. 2011, 286, 2933–2945. [Google Scholar] [CrossRef] [PubMed]
- Thenappan, T.; Ormiston, M.L.; Ryan, J.J.; Archer, S.L. Pulmonary arterial hypertension: Pathogenesis and clinical management. BMJ 2018, 360, j5492. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Li, M.; Wharton, J.; Zhu, M.M.; Zhao, Y.Y. Prolyl-4 Hydroxylase 2 (PHD2) Deficiency in Endothelial Cells and Hematopoietic Cells Induces Obliterative Vascular Remodeling and Severe Pulmonary Arterial Hypertension in Mice and Humans Through Hypoxia-Inducible Factor-2α. Circulation 2016, 133, 2447–2458. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Zhu, M.M.; Peng, Y.; Jin, H.; Machireddy, N.; Qian, Z.; Zhang, X.; Zhao, Y.Y. Endothelial and Smooth Muscle Cell Interaction via FoxM1 Signaling Mediates Vascular Remodeling and Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 788–802. [Google Scholar] [CrossRef]
- Liu, B.; Peng, Y.; Yi, D.; Machireddy, N.; Dong, D.; Ramirez, K.; Dai, J.; Vanderpool, R.; Zhu, M.M.; Dai, Z.; et al. Endothelial PHD2 deficiency induces nitrative stress via suppression of caveolin-1 in pulmonary hypertension. Eur. Respir. J. 2022, 60, 2102643. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.E.; Cober, N.D.; Dai, Z.; Stewart, D.J.; Zhao, Y.Y. Endothelial cells in the pathogenesis of pulmonary arterial hypertension. Eur. Respir. J. 2021, 58, 2003957. [Google Scholar] [CrossRef]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020, 587, 619–625. [Google Scholar] [CrossRef]
- Chan, S.Y.; Rubin, L.J. Metabolic dysfunction in pulmonary hypertension: From basic science to clinical practice. Eur. Respir. Rev. 2017, 26, 170094. [Google Scholar] [CrossRef]
- Xu, W.; Janocha, A.J.; Erzurum, S.C. Metabolism in Pulmonary Hypertension. Annu. Rev. Physiol. 2021, 83, 551–576. [Google Scholar] [CrossRef]
- Shi, X.F.; Su, Y.C. Vascular Metabolic Mechanisms of Pulmonary Hypertension. Curr. Med. Sci. 2020, 40, 444–454. [Google Scholar] [CrossRef]
- Xu, W.; Koeck, T.; Lara, A.R.; Neumann, D.; DiFilippo, F.P.; Koo, M.; Janocha, A.J.; Masri, F.A.; Arroliga, A.C.; Jennings, C.; et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc. Natl. Acad. Sci. USA 2007, 104, 1342–1347. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Erzurum, S.C. Endothelial cell energy metabolism, proliferation, and apoptosis in pulmonary hypertension. Compr. Physiol. 2011, 1, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Zhang, X.; Wang, L.; Yang, Q.; Ma, Q.; Xu, J.; Wang, J.; Kovacs, L.; Ayon, R.J.; Liu, Z.; et al. PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2019, 116, 13394–13403. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Luo, F.; Wu, P.; Huang, Y.; Das, A.; Chen, S.; Chen, J.; Hu, X.; Li, F.; Fang, Z.; et al. Metabolomics reveals metabolite changes of patients with pulmonary arterial hypertension in China. J. Cell Mol. Med. 2020, 24, 2484–2496. [Google Scholar] [CrossRef]
- Umar, S.; Ruffenach, G.; Moazeni, S.; Vaillancourt, M.; Hong, J.; Cunningham, C.; Cao, N.; Navab, S.; Sarji, S.; Li, M.; et al. Involvement of Low-Density Lipoprotein Receptor in the Pathogenesis of Pulmonary Hypertension. J. Am. Heart Assoc. 2020, 9, e012063. [Google Scholar] [CrossRef]
- Singh, N.; Manhas, A.; Kaur, G.; Jagavelu, K.; Hanif, K. Inhibition of fatty acid synthase is protective in pulmonary hypertension. Br. J. Pharmacol. 2016, 173, 2030–2045. [Google Scholar] [CrossRef]
- Hou, C.; Chen, J.; Zhao, Y.; Niu, Y.; Lin, S.; Chen, S.; Zong, Y.; Sun, X.; Xie, L.; Xiao, T. The Emerging Role of Fatty Acid Synthase in Hypoxia-Induced Pulmonary Hypertensive Mouse Energy Metabolism. Oxidative Med. Cell. Longev. 2021, 2021, 9990794. [Google Scholar] [CrossRef]
- Brittain, E.L.; Talati, M.; Fessel, J.P.; Zhu, H.; Penner, N.; Calcutt, M.W.; West, J.D.; Funke, M.; Lewis, G.D.; Gerszten, R.E.; et al. Fatty Acid Metabolic Defects and Right Ventricular Lipotoxicity in Human Pulmonary Arterial Hypertension. Circulation 2016, 133, 1936–1944. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Brittain, E.L.; Trammell, A.W.; Fessel, J.P.; Austin, E.D.; Penner, N.; Maynard, K.B.; Gleaves, L.; Talati, M.; Absi, T.; et al. Evidence for right ventricular lipotoxicity in heritable pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2014, 189, 325–334. [Google Scholar] [CrossRef]
- Zhao, Y.; Peng, J.; Lu, C.; Hsin, M.; Mura, M.; Wu, L.; Chu, L.; Zamel, R.; Machuca, T.; Waddell, T.; et al. Metabolomic heterogeneity of pulmonary arterial hypertension. PLoS ONE 2014, 9, e88727. [Google Scholar] [CrossRef]
- Lee, M.H.; Sanders, L.; Kumar, R.; Hernandez-Saavedra, D.; Yun, X.; Ford, J.A.; Perez, M.J.; Mickael, C.; Gandjeva, A.; Koyanagi, D.E.; et al. Contribution of fatty acid oxidation to the pathogenesis of pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2022, 323, L355–l371. [Google Scholar] [CrossRef] [PubMed]
- Sutendra, G.; Bonnet, S.; Rochefort, G.; Haromy, A.; Folmes, K.D.; Lopaschuk, G.D.; Dyck, J.R.; Michelakis, E.D. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci. Transl. Med. 2010, 2, 44ra58. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Sharma, S.; Fratz, S.; Kumar, S.; Rafikov, R.; Aggarwal, S.; Rafikova, O.; Lu, Q.; Burns, T.; Dasarathy, S.; et al. Disruption of endothelial cell mitochondrial bioenergetics in lambs with increased pulmonary blood flow. Antioxid. Redox Signal. 2013, 18, 1739–1752. [Google Scholar] [CrossRef] [PubMed]
- Fessel, J.P.; Hamid, R.; Wittmann, B.M.; Robinson, L.J.; Blackwell, T.; Tada, Y.; Tanabe, N.; Tatsumi, K.; Hemnes, A.R.; West, J.D. Metabolomic analysis of bone morphogenetic protein receptor type 2 mutations in human pulmonary endothelium reveals widespread metabolic reprogramming. Pulm. Circ. 2012, 2, 201–213. [Google Scholar] [CrossRef]
- Agrawal, V.; Hemnes, A.R.; Shelburne, N.J.; Fortune, N.; Fuentes, J.L.; Colvin, D.; Calcutt, M.W.; Talati, M.; Poovey, E.; West, J.D.; et al. l-Carnitine therapy improves right heart dysfunction through Cpt1-dependent fatty acid oxidation. Pulm. Circ. 2022, 12, e12107. [Google Scholar] [CrossRef]
- Legchenko, E.; Chouvarine, P.; Borchert, P.; Fernandez-Gonzalez, A.; Snay, E.; Meier, M.; Maegel, L.; Mitsialis, S.A.; Rog-Zielinska, E.A.; Kourembanas, S.; et al. PPARγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci. Transl. Med. 2018, 10, eaao0303. [Google Scholar] [CrossRef]
- Fang, Y.H.; Piao, L.; Hong, Z.; Toth, P.T.; Marsboom, G.; Bache-Wiig, P.; Rehman, J.; Archer, S.L. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: Exploiting Randle’s cycle. J. Mol. Med. 2012, 90, 31–43. [Google Scholar] [CrossRef]
- Hassoun, P.M. Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 385, 2361–2376. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Gurtu, V.; Webster, L.; Barnes, G.; Watson, G.; Howard, L.; Cupitt, J.; Paterson, I.; Thompson, R.B.; Chow, K.; et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci. Transl. Med. 2017, 9, eaao4583. [Google Scholar] [CrossRef]
- Michelakis, E.D.; McMurtry, M.S.; Wu, X.C.; Dyck, J.R.; Moudgil, R.; Hopkins, T.A.; Lopaschuk, G.D.; Puttagunta, L.; Waite, R.; Archer, S.L. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: Role of increased expression and activity of voltage-gated potassium channels. Circulation 2002, 105, 244–250. [Google Scholar] [CrossRef]
- McMurtry, M.S.; Bonnet, S.; Wu, X.; Dyck, J.R.; Haromy, A.; Hashimoto, K.; Michelakis, E.D. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ. Res. 2004, 95, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Harvey, L.D.; Chan, S.Y. Emerging Metabolic Therapies in Pulmonary Arterial Hypertension. J. Clin. Med. 2017, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Stone, P.H. Ranolazine: New paradigm for management of myocardial ischemia, myocardial dysfunction, and arrhythmias. Cardiol. Clin. 2008, 26, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.S.; Cuttica, M.J.; Beussink-Nelson, L.; Kozyleva, A.; Sanchez, C.; Mkrdichian, H.; Selvaraj, S.; Dematte, J.E.; Lee, D.C.; Shah, S.J. Effects of ranolazine on exercise capacity, right ventricular indices, and hemodynamic characteristics in pulmonary arterial hypertension: A pilot study. Pulm. Circ. 2015, 5, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Forfia, P.; Vaidya, A.; Mazurek, J.A.; Park, M.H.; Ramani, G.; Chan, S.Y.; Waxman, A.B. Ranolazine Improves Right Ventricular Function in Patients with Precapillary Pulmonary Hypertension: Results From a Double-Blind, Randomized, Placebo-Controlled Trial. J. Card. Fail. 2021, 27, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, R.T.; Hansmann, G.; Snook, S.; Lilienfeld, D.; Rappaport, K.M.; Reaven, G.M.; Rabinovitch, M.; Doyle, R.L. Insulin resistance in pulmonary arterial hypertension. Eur. Respir. J. 2009, 33, 318–324. [Google Scholar] [CrossRef]
- Eurich, D.T.; McAlister, F.A.; Blackburn, D.F.; Majumdar, S.R.; Tsuyuki, R.T.; Varney, J.; Johnson, J.A. Benefits and harms of antidiabetic agents in patients with diabetes and heart failure: Systematic review. BMJ 2007, 335, 497. [Google Scholar] [CrossRef] [PubMed]
- Brittain, E.L.; Niswender, K.; Agrawal, V.; Chen, X.; Fan, R.; Pugh, M.E.; Rice, T.W.; Robbins, I.M.; Song, H.; Thompson, C.; et al. Mechanistic Phase II Clinical Trial of Metformin in Pulmonary Arterial Hypertension. J. Am. Heart Assoc. 2020, 9, e018349. [Google Scholar] [CrossRef]
- Liao, S.; Li, D.; Hui, Z.; McLachlan, C.S.; Zhang, Y. Metformin added to bosentan therapy in patients with pulmonary arterial hypertension associated with congenital heart defects: A pilot study. ERJ Open Res. 2018, 4, 00060–2018. [Google Scholar] [CrossRef]
- Rao, M.S.; Gupta, R.; Liguori, M.J.; Hu, M.; Huang, X.; Mantena, S.R.; Mittelstadt, S.W.; Blomme, E.A.G.; Van Vleet, T.R. Novel Computational Approach to Predict Off-Target Interactions for Small Molecules. Front. Big Data 2019, 2, 25. [Google Scholar] [CrossRef]
- Dai, Z.; Zhao, Y.Y. Discovery of a murine model of clinical PAH: Mission impossible? Trends Cardiovasc. Med. 2017, 27, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Steiner, M.K.; Syrkina, O.L.; Kolliputi, N.; Mark, E.J.; Hales, C.A.; Waxman, A.B. Interleukin-6 overexpression induces pulmonary hypertension. Circ. Res. 2009, 104, 236–244, 228p following 244. [Google Scholar] [CrossRef] [PubMed]
- Dignam, J.P.; Scott, T.E.; Kemp-Harper, B.K.; Hobbs, A.J. Animal models of pulmonary hypertension: Getting to the heart of the problem. Br. J. Pharmacol. 2022, 179, 811–837. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, B.; Dai, Z. Fatty Acid Metabolism in Endothelial Cell. Genes 2022, 13, 2301. https://doi.org/10.3390/genes13122301
Liu B, Dai Z. Fatty Acid Metabolism in Endothelial Cell. Genes. 2022; 13(12):2301. https://doi.org/10.3390/genes13122301
Chicago/Turabian StyleLiu, Bin, and Zhiyu Dai. 2022. "Fatty Acid Metabolism in Endothelial Cell" Genes 13, no. 12: 2301. https://doi.org/10.3390/genes13122301
APA StyleLiu, B., & Dai, Z. (2022). Fatty Acid Metabolism in Endothelial Cell. Genes, 13(12), 2301. https://doi.org/10.3390/genes13122301