
A TARP Syndrome Phenotype Is Associated with a Novel Splicing Variant in RBM10


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
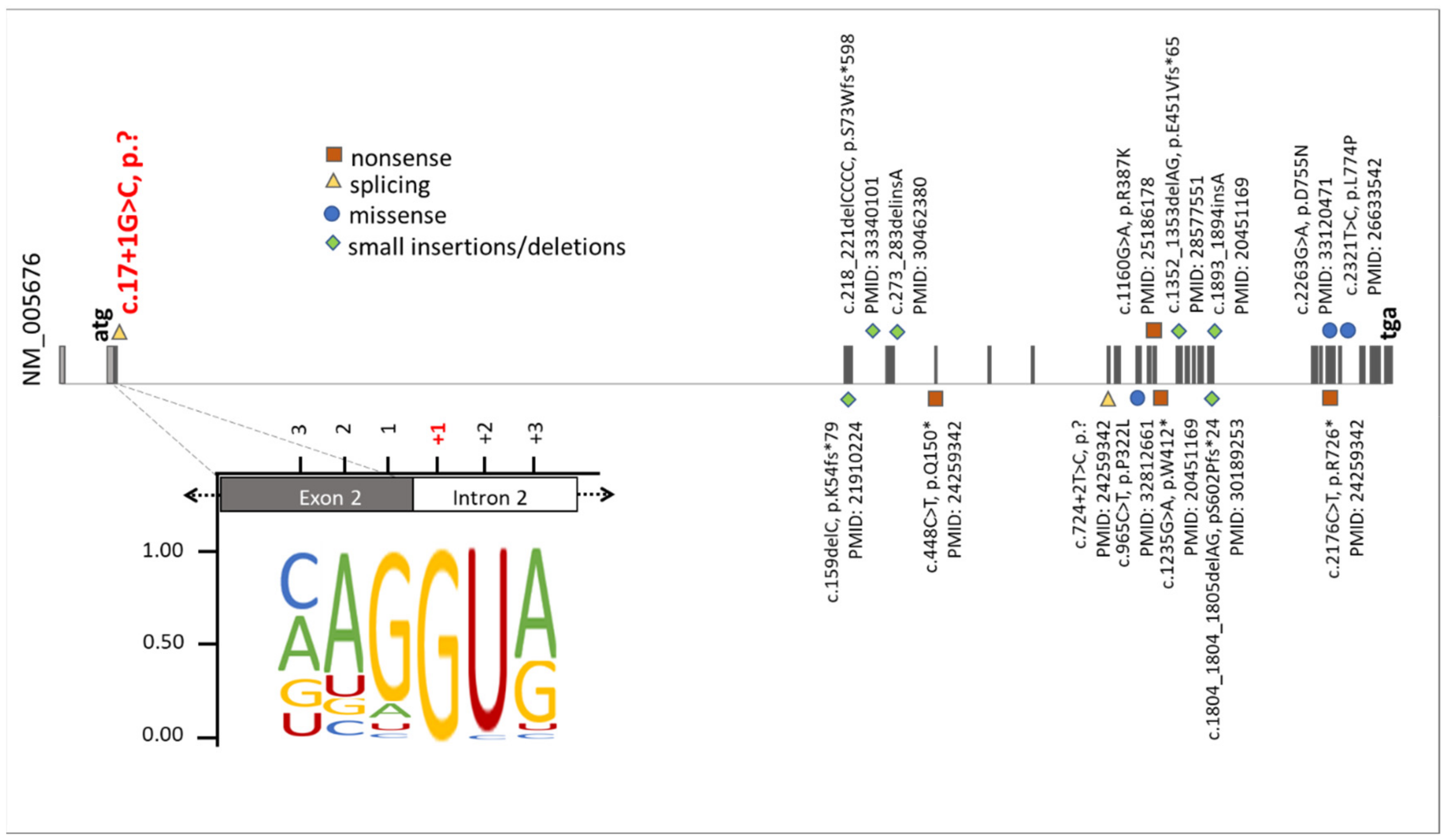
1. Introduction
2. Material and Methods
2.1. Probands and Ethical Compliance
2.2. DNA Extraction
2.3. Whole Exome Sequencing (WES) and Computer Analyses
2.4. Primers, PCR Amplification and Sanger Sequencing
2.5. Cell Culture of Patient-Derived Fibroblasts
2.6. RNA Isolation, cDNA, and Reverse Transcriptase PCR (RT-PCR)
2.7. Immunocytochemical Staining (ICC)
2.8. Western Blotting (WB)
3. Results
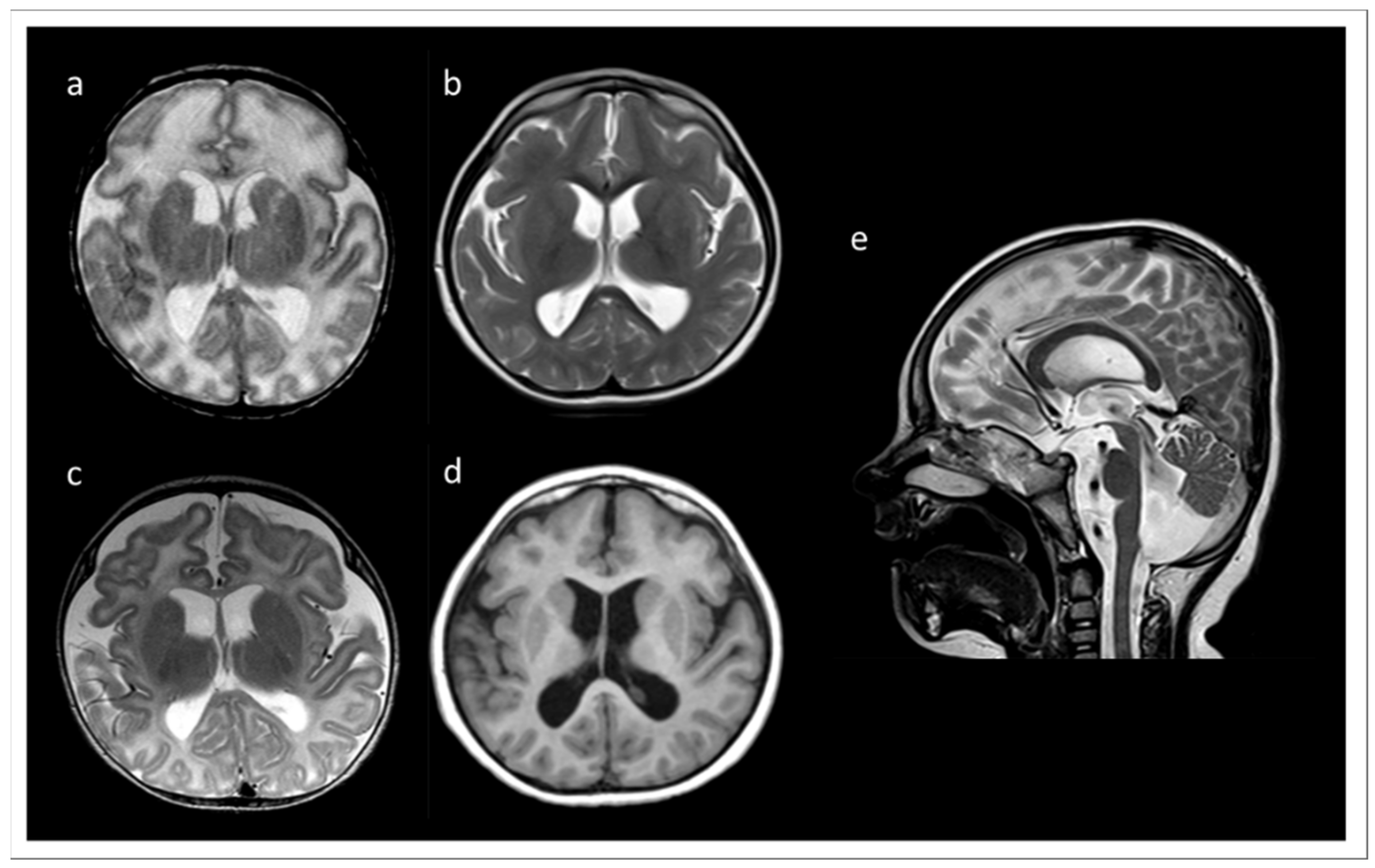
3.1. Clinical Data and Initial Routine Genetic Diagnostics
3.2. Routine Cytogenetics and Molecular Gene Testing
3.3. WES Outcome
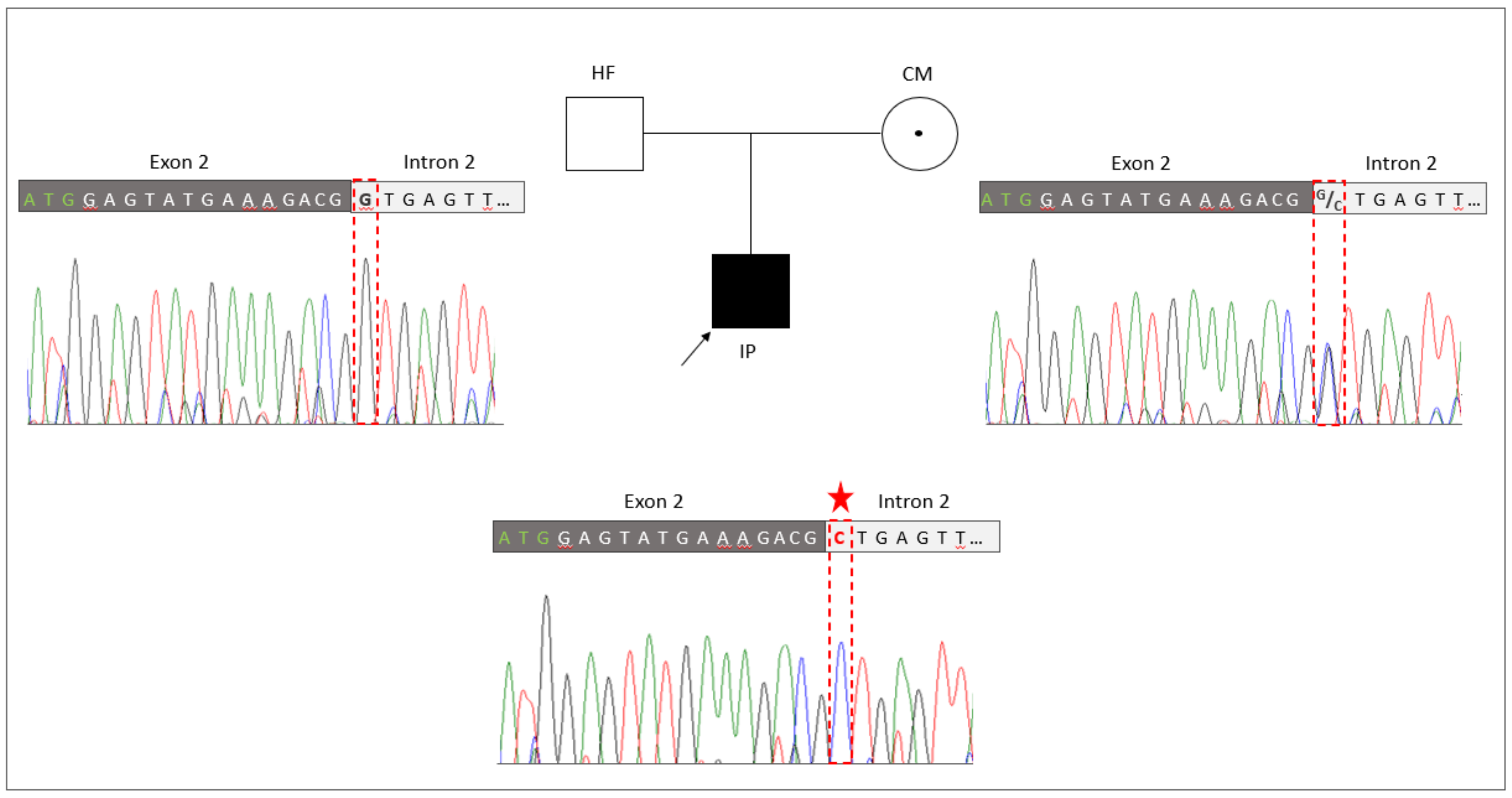
3.4. Familiar Genotyping Analyses
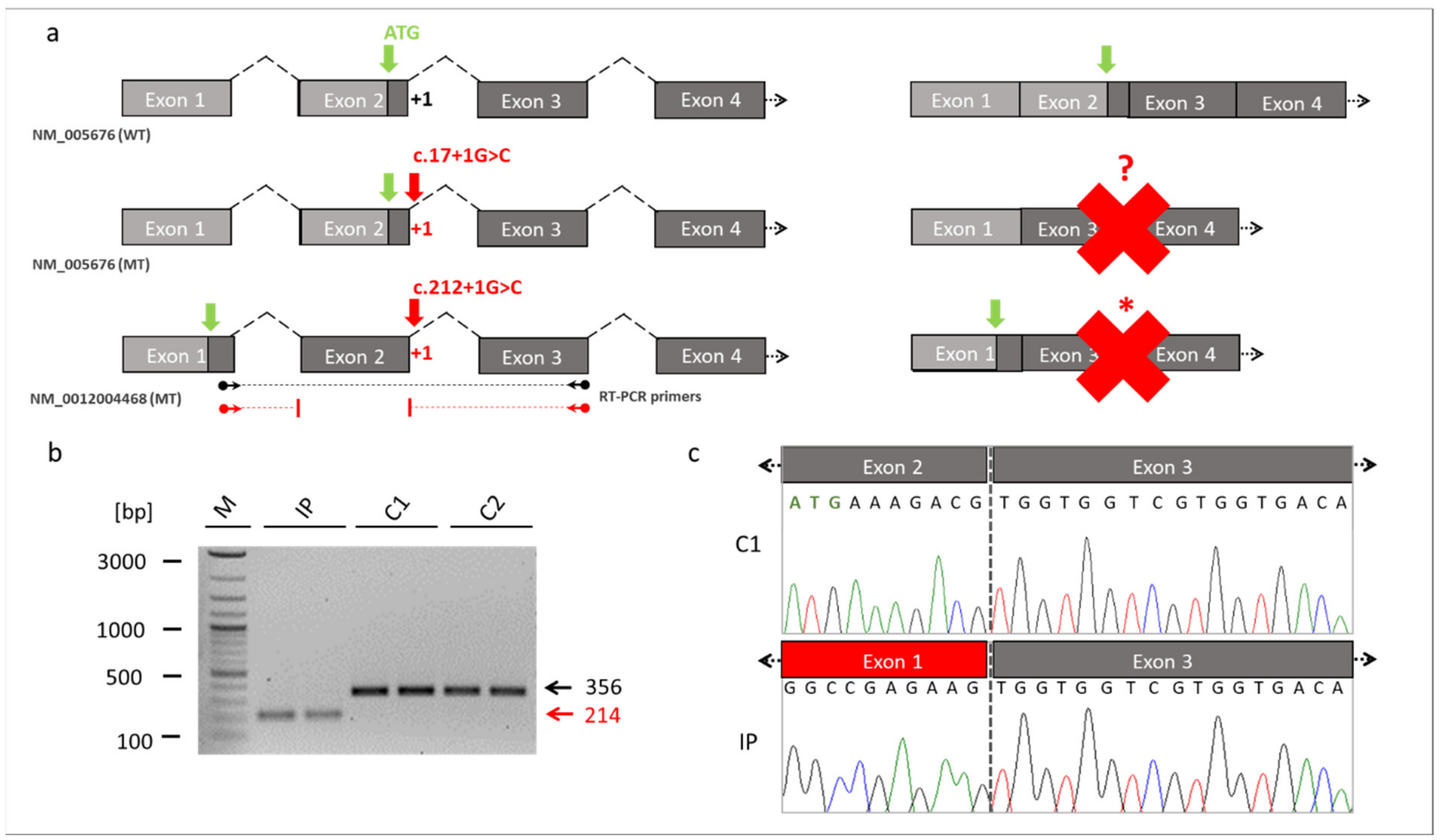
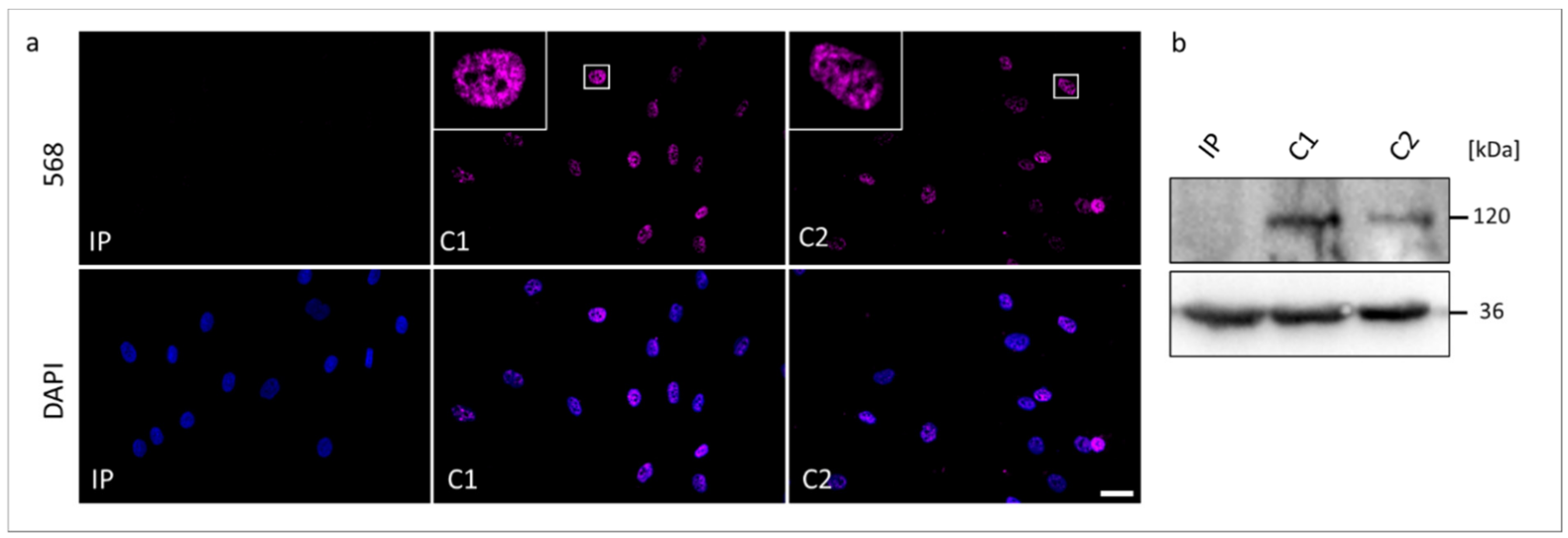
3.5. Transcript and Protein Analyses (RT-PCR, ICC, and WB)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kurpinski, K.T.; Magyari, P.A.; Gorlin, R.J.; Ng, D.; Biesecker, L.G. Designation of the TARP syndrome and linkage to Xp11.23-q13.3 without samples from affected patients. Am. J. Med. Genet. A 2003, 120A, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Gorlin, R.J.; Cervenka, J.; Anderson, R.C.; Sauk, J.J.; Bevis, W.D. Robin’s syndrome. A probably X-linked recessive subvariety exhibiting persistence of left superior vena cava and atrial septal defect. Am. J. Dis. Child. 1970, 119, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.J.; Sapp, J.C.; Curry, C.; Horton, M.; Leon, E.; Cusmano-Ozog, K.; Dobyns, W.B.; Hudgins, L.; Zackai, E.; Biesecker, L.G. Expansion of the TARP syndrome phenotype associated with de novo mutations and mosaicism. Am. J. Med. Genet. A 2014, 164A, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Kaeppler, K.E.; Stetson, R.C.; Lanpher, B.C.; Collura, C.A. Infant male with TARP syndrome: Review of clinical features, prognosis, and commonalities with previously reported patients. Am. J. Med. Genet. A 2018, 176, 2911–2914. [Google Scholar] [CrossRef]
- Powis, Z.; Hart, A.; Cherny, S.; Petrik, I.; Palmaer, E.; Tang, S.; Jones, C. Clinical diagnostic exome evaluation for an infant with a lethal disorder: Genetic diagnosis of TARP syndrome and expansion of the phenotype in a patient with a newly reported RBM10 alteration. BMC Med. Genet. 2017, 18, 60. [Google Scholar] [CrossRef]
- Furukawa, T.; Kuboki, Y.; Tanji, E.; Yoshida, S.; Hatori, T.; Yamamoto, M.; Shibata, N.; Shimizu, K.; Kamatani, N.; Shiratori, K. Whole-exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Sci. Rep. 2011, 1, 161. [Google Scholar] [CrossRef]
- Gripp, K.W.; Hopkins, E.; Johnston, J.J.; Krause, C.; Dobyns, W.B.; Biesecker, L.G. Long-term survival in TARP syndrome and confirmation of RBM10 as the disease-causing gene. Am. J. Med. Genet. A 2011, 155A, 2516–2520. [Google Scholar] [CrossRef]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef]
- Johnston, J.J.; Teer, J.K.; Cherukuri, P.F.; Hansen, N.F.; Loftus, S.K.; Center, N.I.H.I.S.; Chong, K.; Mullikin, J.C.; Biesecker, L.G. Massively parallel sequencing of exons on the X chromosome identifies RBM10 as the gene that causes a syndromic form of cleft palate. Am. J. Hum. Genet. 2010, 86, 743–748. [Google Scholar] [CrossRef]
- Hernandez, J.; Bechara, E.; Schlesinger, D.; Delgado, J.; Serrano, L.; Valcarcel, J. Tumor suppressor properties of the splicing regulatory factor RBM10. RNA Biol. 2016, 13, 466–472. [Google Scholar] [CrossRef]
- Mueller, C.F.; Berger, A.; Zimmer, S.; Tiyerili, V.; Nickenig, G. The heterogenous nuclear riboprotein S1-1 regulates AT1 receptor gene expression via transcriptional and posttranscriptional mechanisms. Arch. Biochem. Biophys. 2009, 488, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, L.C.; Rintala-Maki, N.D.; White, R.D.; Morin, C.D. RNA binding motif (RBM) proteins: A novel family of apoptosis modulators? J. Cell Biochem. 2005, 94, 5–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gogol-Doring, A.; Hu, H.; Frohler, S.; Ma, Y.; Jens, M.; Maaskola, J.; Murakawa, Y.; Quedenau, C.; Landthaler, M.; et al. Integrative analysis revealed the molecular mechanism underlying RBM10-mediated splicing regulation. EMBO Mol. Med. 2013, 5, 1431–1442. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Yamamoto, N.; Kimura, M.; Nishio, K.; Yamane, H.; Nakajima, K. RBM10 regulates alternative splicing. FEBS Lett. 2014, 588, 942–947. [Google Scholar] [CrossRef]
- Weigt, M.; Gao, Q.; Ban, H.; He, H.; Mastrobuoni, G.; Kempa, S.; Chen, W.; Li, F. Rbm10 facilitates heterochromatin assembly via the Clr6 HDAC complex. Epigenetics Chromatin. 2021, 14, 8. [Google Scholar] [CrossRef]
- Garcia-Blanco, M.A.; Baraniak, A.P.; Lasda, E.L. Alternative splicing in disease and therapy. Nat. Biotechnol. 2004, 22, 535–546. [Google Scholar] [CrossRef]
- Padgett, R.A. New connections between splicing and human disease. Trends Genet. 2012, 28, 147–154. [Google Scholar] [CrossRef]
- Tazi, J.; Bakkour, N.; Stamm, S. Alternative splicing and disease. Biochim. Biophys. Acta. 2009, 1792, 14–26. [Google Scholar] [CrossRef]
- Kumps, C.; D’Haenens, E.; Vergult, S.; Leus, J.; van Coster, R.; Jansen, A.; Devriendt, K.; Oostra, A.; Vanakker, O.M. Phenotypic spectrum of the RBM10-mediated intellectual disability and congenital malformation syndrome beyond classic TARP syndrome features. Clin. Genet. 2021, 99, 449–456. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A. Primer3Plus, an enhanced web interface to Primer3. Nucleic. Acids. Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef] [PubMed]
- Reiff, C.; Owczarek-Lipska, M.; Spital, G.; Roger, C.; Hinz, H.; Juschke, C.; Thiele, H.; Altmuller, J.; Nurnberg, P.; Da Costa, R.; et al. The mutation p.E113K in the Schiff base counterion of rhodopsin is associated with two distinct retinal phenotypes within the same family. Sci. Rep. 2016, 6, 36208. [Google Scholar] [CrossRef] [PubMed]
- Glaus, E.; Schmid, F.; Da Costa, R.; Berger, W.; Neidhardt, J. Gene therapeutic approach using mutation-adapted U1 snRNA to correct a RPGR splice defect in patient-derived cells. Mol. Ther. 2011, 19, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Villegas, J.; McPhaul, M. Establishment and culture of human skin fibroblasts. Curr. Protoc. Mol. Biol. 2005, 71, 28.3.1–28.3.9. [Google Scholar] [CrossRef] [PubMed]
- Markus, F.; Kannengiesser, A.; Nader, P.; Atigbire, P.; Scholten, A.; Vossing, C.; Bultmann, E.; Korenke, G.C.; Owczarek-Lipska, M.; Neidhardt, J. A novel missense variant in the EML1 gene associated with bilateral ribbon-like subcortical heterotopia leads to ciliary defects. J. Hum. Genet. 2021, 66, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Wachutka, L.; Caizzi, L.; Gagneur, J.; Cramer, P. Global donor and acceptor splicing site kinetics in human cells. eLife 2019, 8. [Google Scholar] [CrossRef]
- Hojland, A.T.; Lolas, I.; Okkels, H.; Lautrup, C.K.; Diness, B.R.; Petersen, M.B.; Nielsen, I.K. First reported adult patient with TARP syndrome: A case report. Am. J. Med. Genet. A 2018, 176, 2915–2918. [Google Scholar] [CrossRef]
- Imagawa, E.; Konuma, T.; Cork, E.E.; Diaz, G.A.; Oishi, K. A novel missense variant in RBM10 can cause a mild form of TARP syndrome with developmental delay and dysmorphic features. Clin. Genet. 2020, 98, 606–612. [Google Scholar] [CrossRef]
- Anna, A.; Monika, G. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef]
- Lenaers, G.; Neutzner, A.; Le Dantec, Y.; Juschke, C.; Xiao, T.; Decembrini, S.; Swirski, S.; Kieninger, S.; Agca, C.; Kim, U.S.; et al. Dominant optic atrophy: Culprit mitochondria in the optic nerve. Prog. Retin. Eye. Res. 2021, 83, 100935. [Google Scholar] [CrossRef]
- Schmid, F.; Glaus, E.; Barthelmes, D.; Fliegauf, M.; Gaspar, H.; Nurnberg, G.; Nurnberg, P.; Omran, H.; Berger, W.; Neidhardt, J. U1 snRNA-mediated gene therapeutic correction of splice defects caused by an exceptionally mild BBS mutation. Hum. Mutat. 2011, 32, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Baralle, D.; Baralle, M. Splicing in action: Assessing disease causing sequence changes. J. Med. Genet. 2005, 42, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Lord, J.; Baralle, D. Splicing in the Diagnosis of Rare Disease: Advances and Challenges. Front. Genet. 2021, 12, 689892. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.G.; Smith, C.W.J. Intron retention as a component of regulated gene expression programs. Hum. Genet. 2017, 136, 1043–1057. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Ding, D.; Li, X.; Shen, T.; Fu, H.; Zhong, H.; Wei, G.; Ni, T. Prevalent intron retention fine-tunes gene expression and contributes to cellular senescence. Aging Cell 2020, 19, e13276. [Google Scholar] [CrossRef] [PubMed]
- Mauger, O.; Scheiffele, P. Beyond proteome diversity: Alternative splicing as a regulator of neuronal transcript dynamics. Curr. Opin. Neurobiol. 2017, 45, 162–168. [Google Scholar] [CrossRef]
- Waterham, H.R.; Koster, J.; van Roermund, C.W.; Mooyer, P.A.; Wanders, R.J.; Leonard, J.V. A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 2007, 356, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- Sayer, J.A.; Otto, E.A.; O’Toole, J.F.; Nurnberg, G.; Kennedy, M.A.; Becker, C.; Hennies, H.C.; Helou, J.; Attanasio, M.; Fausett, B.V.; et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat. Genet. 2006, 38, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Najm, J.; Horn, D.; Wimplinger, I.; Golden, J.A.; Chizhikov, V.V.; Sudi, J.; Christian, S.L.; Ullmann, R.; Kuechler, A.; Haas, C.A.; et al. Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat. Genet. 2008, 40, 1065–1067. [Google Scholar] [CrossRef]
- Maydan, G.; Noyman, I.; Har-Zahav, A.; Neriah, Z.B.; Pasmanik-Chor, M.; Yeheskel, A.; Albin-Kaplanski, A.; Maya, I.; Magal, N.; Birk, E.; et al. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J. Med. Genet. 2011, 48, 383–389. [Google Scholar] [CrossRef]
- McMillin, M.J.; Below, J.E.; Shively, K.M.; Beck, A.E.; Gildersleeve, H.I.; Pinner, J.; Gogola, G.R.; Hecht, J.T.; Grange, D.K.; Harris, D.J.; et al. Mutations in ECEL1 cause distal arthrogryposis type 5D. Am. J. Hum. Genet. 2013, 92, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Axelrod, L.; Whitcomb, R.W.; Harris, P.E.; Crowley, W.F.; Jameson, J.L. Hypogonadism caused by a single amino acid substitution in the beta subunit of luteinizing hormone. N. Engl. J. Med. 1992, 326, 179–183. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Owczarek-Lipska, M.; Markus, F.; Bültmann, E.; Korenke, G.C.; Neidhardt, J. A TARP Syndrome Phenotype Is Associated with a Novel Splicing Variant in RBM10. Genes 2022, 13, 2154. https://doi.org/10.3390/genes13112154
Owczarek-Lipska M, Markus F, Bültmann E, Korenke GC, Neidhardt J. A TARP Syndrome Phenotype Is Associated with a Novel Splicing Variant in RBM10. Genes. 2022; 13(11):2154. https://doi.org/10.3390/genes13112154
Chicago/Turabian StyleOwczarek-Lipska, Marta, Fenja Markus, Eva Bültmann, G. Christoph Korenke, and John Neidhardt. 2022. "A TARP Syndrome Phenotype Is Associated with a Novel Splicing Variant in RBM10" Genes 13, no. 11: 2154. https://doi.org/10.3390/genes13112154
APA StyleOwczarek-Lipska, M., Markus, F., Bültmann, E., Korenke, G. C., & Neidhardt, J. (2022). A TARP Syndrome Phenotype Is Associated with a Novel Splicing Variant in RBM10. Genes, 13(11), 2154. https://doi.org/10.3390/genes13112154