Abstract
The spotted catfish, Arius maculatus (Siluriformes), is an important economical aquaculture species inhabiting the Indian Ocean, as well as the western Pacific Ocean. The bioinformatics data in previous studies about the phylogenetic reconstruction of Siluriformes were insufficient and incomplete. In the present study, we presented a newly sequenced A. maculatus mitochondrial genome (mtDNA). The A. maculatus mtDNA was 16,710 bp in length and contained two ribosomal RNA (rRNA) genes, thirteen protein-coding genes (PCGs), twenty-two transfer RNA (tRNA) genes, and one D-loop region. The composition and order of these above genes were similar to those found in most other vertebrates. The relative synonymous codon usage (RSCU) of the 13 PCGs in A. maculatus mtDNA was consistent with that of PCGs in other published Siluriformes mtDNA. Furthermore, the average non-synonymous/synonymous mutation ratio (Ka/Ks) analysis, based on the 13 PCGs of the four Ariidae species, showed a strong purifying selection. Additionally, phylogenetic analysis, according to 13 concatenated PCG nucleotide and amino acid datasets, showed that A. maculatus and Netuma thalassina (Netuma), Occidentarius platypogon (Occidentarius), and Bagre panamensis (Bagre) were clustered as sister clade. The complete mtDNA of A. maculatus provides a helpful dataset for research on the population structure and genetic diversity of Ariidae species.
1. Introduction
In metazoans, the typical complete mitochondrial genome (mtDNA) is usually a circular, double-stranded molecule, with sizes ranging from 13 to 20 kb, containing 13 protein-coding genes (PCGs), 2 ribosomal RNA (rRNA) genes, and 22 transfer RNA (tRNA) genes. Moreover, an A + T-rich region, which includes the initial sites for RNA transcription and mtDNA replication, is regarded as the non-coding region or the control region (CR) [1]. Owing to its maternal inheritance, rapid evolutionary rate, short coalescence time, conserved gene content, small genome size, and low levels of sequence recombination [2,3], mtDNA is widely used in various research fields, such as species identification and taxonomic resolution [4,5], comparative and molecular evolution [6,7,8], population genetics [9], and non-synonymous (Ka) and synonymous ㉿ substitutions of many species [5,10,11,12].
Moreover, mtDNA is commonly known as a helpful molecular marker for phylogenetic analyses among fish taxa. A single mitochondrial gene fragment has limitations in resolving complex phylogenetic relationships in plentiful fish lineages [13]. The additional informative sites from complete mtDNA allow these detailed branches and higher-level relationships to be more adequately resolved [14]. Consequently, in the present study, the mtDNA data may improve the understanding of the evolutionary relationships of the family Siluriformes.
Arius maculatus, also known as the spotted catfish, belongs to Ariidae, Siluriformes. It is a benthic species in subtropical and tropical waters, inhabiting the bottoms of rivers, estuaries, and coasts, and is extensively distributed in the Indo-West Pacific (http://fishbase.sinica.edu.tw/Summary/SpeciesSummary.php, accessed on 4 March 2019) [15]. It has a long body shape, broad front, lateral flat rear, and a body length of more than 60 cm. Furthermore, there are serrated poison glands at the base of the dorsal and pectoral spines, which cause severe pain when stabbed, and are the defensive tools of the fish. The fish has a strong smell, but it has a high fat content. Southeast coastal residents commonly cook it with “angelica” and other traditional Chinese medicine. The feeding strategy, morphology, and ecology of this important species has been studied in recent years [16].
A better understanding of Siluriformes mtDNAs requires expanded taxon sampling. Siluriformes includes approximately 3093 described species, classified into 478 genera and 36 superfamilies [17]. Ariidae includes approximately 26 genera and over 133 species [17,18], many of which are agriculturally important. The classification of Ariidae species is arguably the most poorly resolved of any catfish family [19]. Simply, the subfamilial divisions within the Ariidae (Galeichthyinae and Ariinae) were absolutely consistent among the four reconstruction methods conducted (MP, BI, ML-RAxML, and ML-Garli) and well-supported [20]. Moreover, mtDNA synteny analysis has revealed many common mtDNA features in Siluriformes, which may lead to a better understanding of the evolution of Siluriformes [21]. Despite the vast species diversity in this family, there are only five species containing available complete mtDNAs in the GenBank database, and mtDNA information in the family Ariidae is only available for three species, Occidentarius platypogon [22], Bagre panamensis [23], and Arius arius [24]. To date, there is still an observable lack of mtDNA information among Ariidae, and the phylogenetic relationships and taxonomic status of A. maculatus in Ariidae are still vague. Consequently, to understand the evolutionary relationships of A. maculatus in Siluriformes and further study the population genetics in Ariidae, we sequenced the complete mtDNA of A. maculatus and analyzed its characteristics and evolutionary relationships.
2. Materials and Methods
2.1. Samples and Mitogenome Sequencing
Adult A. maculatus fish (about 2400 g) was obtained from Beibu Gulf, China (longitude 21°36′50″ N and latitude 108°44′00″ E) in June 2019 and directly frozen. Genomic DNA was extracted from muscle tissues, according to the instructions of Genomic DNA Extraction Ver.5.0 kit (TaKaRa, Kyoto, Japan). The concentration of the isolated gDNA was detected using the NANODROP 2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA). The quality of the extracted gDNA was evaluated by electrophoresis with 1% agarose gel and stained with Gel Red™ (Biotium). Then, normalized genomic DNA (4 μg) was used to prepare the paired-end library, according to the instructions of the NEBNext DNA sample libraries kit (NEB, New England). The quantification and size of the library was estimated using a Bioanalyzer 2100 High Sensitivity DNA chip (Agilent, CA, USA). Sequencing of the normalized library (2 nM) was performed on a HiSeq 2500 platform (2 × 101 bp paired-end reads) (San Diego, IL, USA).
2.2. Genome Assembly and Annotation
Clean data were generated according to a previous protocol [25], and the remaining high-quality reads were then assembled using SeqMan NGen (http://www.dnastar.com/t-tutorials-seqman-ngen.aspx, accessed on 10 March 2021) (DNASTAR Inc., Madison, WI, USA). Match spacing, minimum match percentage, match size, gap penalty, mismatch penalty, expected genome length, and maximum gap length were set to 10, 93, 50, 30, 20, 16,000, and 6%, respectively. After alignment with the NCBI nt database, the sequences were aligned using the blastn method (https://blast.ncbi.nlm.nih.gov/, accessed on 10 March 2021). Siluriformes mtDNA-mapped sequences were identified as A. maculatus mtDNA. To demonstrate the preciseness of the assembled genome sequence, primers were used to amplify mtDNA (Table S1). PCR amplification has been previously described [4]. Moreover, PCGs, rRNA genes, tRNA genes, and the D-loop region of mtDNA were annotated by the Mito Annotator (http://mitofish.aori.u-tokyo.ac.jp/annotation/input.html, accessed on 8 May 2021), according to circular genome parameters [26].
2.3. Genome Sequence Analysis
To confirm tRNAs, the tRNAscan-SE Search Server 1.21 program was used [27,28]. OGDRAW1.2 was used to create the gene map of A. maculatus mtDNA, and hand annotation was completed [29]. An estimate of strand skew was developed using a previous study’s formulae [30]. By using “models- > Compute Codon Usage Bias” in MEGA 6.0, relative synonymous codon usage (RSCU) was calculated [31]. The nonsynonymous/synonymous mutation (Ka/Ks) ratio and codon usage in the 13 PCGs were calculated using DnaSP 5.10.01 to investigate the evolutionary branching of the Ariidae lineage [32]. In addition, we determined the skew of AT and GC in the whole mtDNA, PCG, tRNA, rRNA, or control region sequence, using the following formula: AT skew = (A − T) /(A + T) and GC skew = (G − C)/ (G + C) [33].
2.4. Phylogenetic Analysis
Phylogenetic analysis of Siluriformes was carried out using 13 PCG nucleotide and amino acid sequences from 21 species. Based on MUSCLE v.3.8.31, each of the 13 PCG nucleotide and amino acid sequences from all 21 species was individually aligned (http://www.drive5.com/muscle/, accessed on 8 May 2021) [34] and then aggregated into a sequence matrix to reconstruct the phylogeny. The 21 mitogenome data were all downloaded from the NCBI database. Twenty-one species were divided into 15 genera and 7 families in the order Siluriformes. To test the nucleic acid and amino acid models, we used jModelTest2.1.7 (https://code.google.com/p/jmodeltest2/, accessed on 8 May 2021) [35] and Prottest3.2 [36]. Akaike information criterion(AIC), was considered the best model for tree formation. The maximum likelihood (ML) tree was implemented in RAxML 8.0.12 [37] under the GTR-γ model and MtMam+I+G model for nucleic acid and amino acid trees, respectively, and node support was calculated with 1000 bootstrap replications (random seed value of 1,234,567). Further, MrBayes 3.2.5 [38] for Bayesian inference (BI) was used to reconstruct phylogenetic trees with 10,000,000 generations. The BI analysis used the CAT-GTR model, and two independent Markov chain Monte Carlo (MCMC) chains were run for 10,000 cycles. Phylogenetic trees were generated using FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/, accessed on 8 May 2021).
3. Results and Discussion
3.1. Genome Size and Organization
Raw data of approximately 1.5 G with read lengths of 150 bp were generated. The mtDNA sequences covered 100% of the genome and were approximately 57X deep. The total number of bases (bp) was 965,100, and the read number was 6434. The whole mtDNA of Arius maculatus was a circular double-chain DNA molecule with a length of 16,710 bp (GenBank: MN604079; Figure 1, Table 1), which is comparable to the mtDNA of other Siluriformes species, ranging from 16,471 bp (Pangasius larnaudii) to 16,830 bp (Ariopsis seemanni) [39] (Table S2). Nucleotide BLAST of the complete A. maculatus mtDNA against other Siluriformes mtDNA showed sequence homology of 99.80 (N. thalassina), 99.74 (A. arius), 90.31 (A. seemanni), and 90.14% (O. platypogon) with closely related species, and of 83.25 (Glaridoglanis andersonii), 83.18 (Silurus soldatovi), 83.15 (S. meridionalis), and 83.12% (Silurus asotus) with distantly related species (Table S2). Moreover, the mtDNA of A. maculatus comprised 2 rRNA genes, 13 PCGs, 22 tRNA genes, and a D-loop region. The arrangement of the genes in the A. maculatus mtDNA was identical to that of other reported Ariidae mtDNAs (Table 1) [22,23,24]. Of these genes, 29 (12 PCG, 2 rRNA, and 15 tRNA) were located in the heavy strand (H-strand); the rest (1 PCG and 8 tRNA) were located in the light strand (L-strand) (Table 1). As valid species markers and genus authentication features, these typical features have also been observed in other Siluriformes [39,40,41,42].
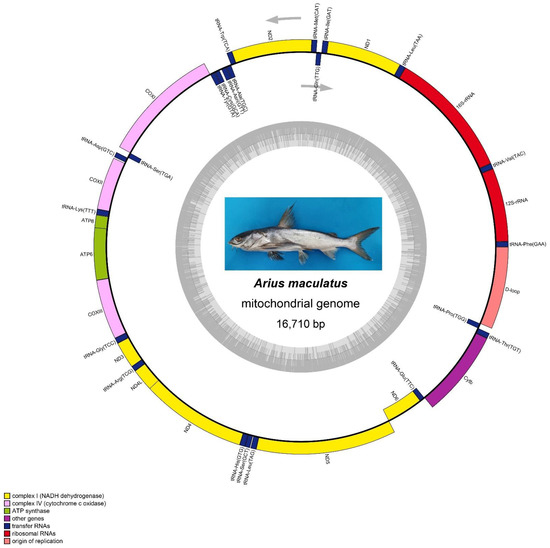
Figure 1.
Map of the Arius maculatus mtDNA. Image of the A. maculatus was shown in the middle. The genes inside are transcribed counterclockwise, whereas the genes outside the circle are transcribed clockwise. Gene blocks are filled with different colors, as shown in the cutline. The inner ring shadow indicates the GC content of the mtDNA.

Table 1.
Sequence characteristics of Arius maculatus mitochondrial genome.
The nucleotide composition of the mtDNA was A (29.63%), T (25.42%), C (29.65%), and G (15.30%), with a high A + T nucleotide content (55.05%), which was 54.88, 55.52, 52.75, and 62.55% for the PCGs, tRNAs, rRNAs, and D-loop region, respectively (Table 2). Among Siluriformes, A. maculatus had the lowest A + T nucleotide composition. With more As than Ts, the AT skew (0.0764) observed here was similar to that in O. platypogon (0.0765), N. thalassina (0.0775), and B. panamensis (0.0716), which are evolutionarily closely related. Most Siluriformes, however, exhibited a positive AT skew in their mtDNA (Table 2). GC skews ranged from −0.3308 in O. platypogon to −0.2799 in S. soldatovi (Table 2). In A. maculatus, the mtDNA was negative (−0.3194), showing that it had a GC skew more toward Cs than Gs.
Table 2.
Nucleotide composition of the mitochondrial genome in different Siluriformes.
3.2. Protein-Coding Gene Features
The PCG sequences make up 68.26% of the A. maculatus mtDNA, with 11,407 bp. Furthermore, 19 Siluriformes were shown to have AT skews and GC skews that differed from nucleotide composition (Table 2). Among Siluriformes species, A. maculatus mtDNA had a moderate AT skew value (0.0489) of the PCG region. Other species, however, showed a negative GC skew (−0.3888) [43,44,45]. In addition, thirteen PCGs with AT and GC skews were also calculated in Figure S1, indicating that they were mutually consistent and closely related [43,45].
With the exception of COXI, which starts with a GTG codon, each PCG is initiated by a classic ATN codon (Table 1). Other Ariidae fish have shown similar results. In six of the thirteen PCGs (ND1, COXI, ATP8, ATP6, ND4L, and ND5), a typical termination codon (TAA) is used, which is common to Siluriformes mtDNA [19,21]. COXII, COXIII, ND4, and Cytb, on the other hand, terminate with the incomplete termination signal T, whereas ND2, ND3, and ND6 terminate with TAG. (Table 1). The mtDNA of A. maculatus is homologous to sequenced mtDNAs of other Siluriformes, including A. arius [24], O. platypogon [24], Pseudecheneis immaculatus [46], and Ailia coila [47].
A. maculatus encodes 3792 amino acids in its 13 PCGs. Moreover, codon usage is displayed in Table 3. A. maculatus PCGs were dominated by the following amino acids: leucine (Leu, 17.1%), alanine (Ala, 8.84%), isoleucine (Ile, 8.07%), and threonine (Thr, 7.68%), whereas those encoding cysteine (Cys, 0.71%) were rare (Table 3). RSCU analysis of the 13 PCGs indicated that the codons encoding Leu and serine (Ser) were most abundant in A. maculatus (Figure 2). In the PCGs of the eight species examined, there was homology in amino acid content and codon distribution between those species (Figure 3). It was deduced that conserved amino acid sequences were found in Siluriformes [39,42,48]. Furthermore, A or T in the third position was overused compared with other synonymous codons [4]. For example, codons for leucine (TTG) and serine (TCG) were rare, whereas CTA and TCA were widespread (Figure 4).
Table 3.
Codon usage of A. maculatus mitochondrial PCGs.
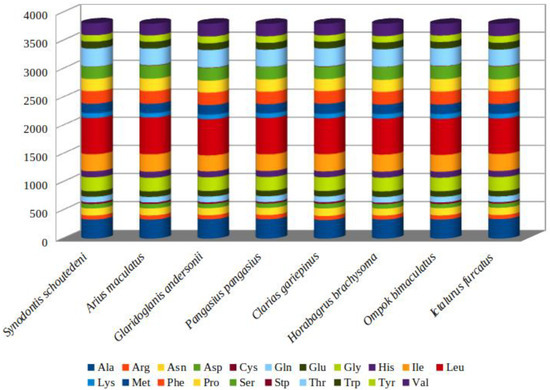
Figure 2.
Comparison of codon usage within the mtDNA of members of the Siluriformes. Species (A. maculatus, Pangasius, Horabagrus brachysoma, Synodontis schoutedeni, Clarias gariepinus, Ictalurus furcatus, Ompok bimaculatus, and Glaridoglanis andersonii) represent the superfamily to which the species belongs (Ariidae, Pangasiidae, Bagridae, Mochokinae, Clariidae, Ictaluridae, Siluridae, and Sisoridae).
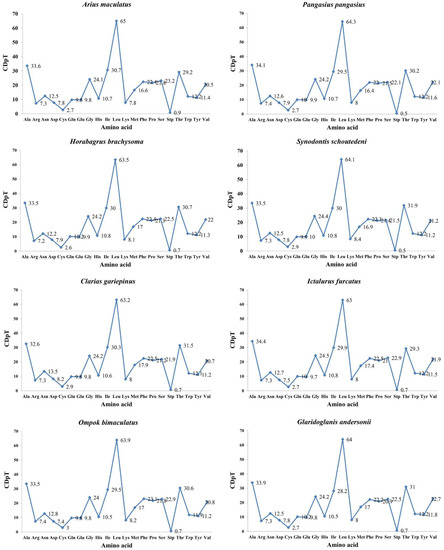
Figure 3.
Codon distribution in members of eight superfamilies in the Siluriformes. CDspT = codons per thousand codons.
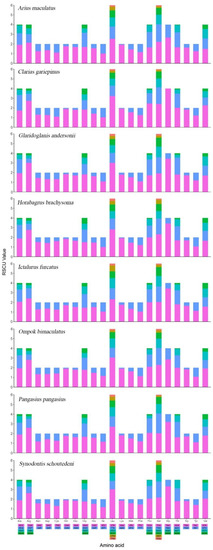
Figure 4.
Relative synonymous codon usage (RSCU) of the mtDNA of 8 superfamilies in the Siluriformes. Codon families are plotted on the x-axis. Codons indicated above the bar are not present in the mtDNA.
3.3. Transfer RNAs and Ribosomal RNAs
Twenty-two classical structures of tRNAs were validated in A. maculatus mtDNA, with lengths ranging from 66 (tRNACys) to 75 bp (tRNALeu), with a total length of 1558 bp (Table 1). The lowest A + T content of tRNAs was found in A. maculatus (55.52%), N. thalassina (55.52%), and S. soldatovi (55.44%), and the highest was found in Mystus cavasius (55.84%). (Table 2). On the H strand, there were fourteen tRNA genes encoded, whereas the residues are on the L strand indicated that there were four tRNA genes (Table 1). The AT (0.1283) and GC skews (−0.1631) of A. maculatus were similar to those of several sequenced Siluriformes mtDNAs, such as O. platypogon and N. thalassina (Table 2). The predicted tRNAs are shown in Figure 5. Except for tRNASer (GCT), which lacks the dihydrouridine ‘DHU’ arm, all tRNAs formed typical clover-leaf secondary structures in A. maculatus (Figure 5). The tRNASer ‘DHU’ arm is a large loop substitute for the conserved stem-and-loop structure. This representative characteristic [1] was also observed in the mtDNA of other Siluriformes species, including Ompok bimaculatus [48], Hemibagrus sp. [49], Silurus lanzhouensis [50], and Chrysichthys nigrodigitatus [51]. Twelve tRNA genes had at least one G-T mismatch, which caused a weak bond. In the amino acid acceptor stems of tRNACys (GCA) and tRNAMet (CAT), five T-T mismatches were observed. (Figure 5). It was also found that tRNALeu (TAA) contained an A-G mismatch. In tRNA sequences, the RNA-editing mechanism, well-known in vertebrate mtDNA, can correct unmatched base pairs [52].
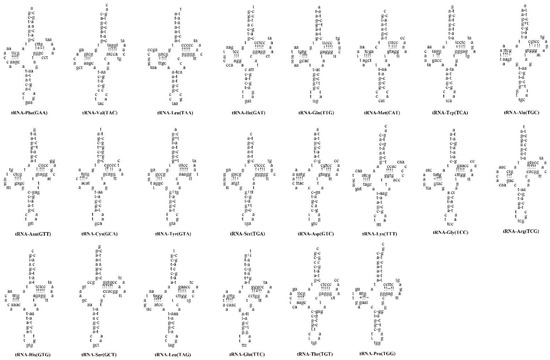
Figure 5.
Putative secondary structures for 22 tRNA genes in mtDNA of A. maculatus. Watson–Crick and GT bonds are expounded by “-” and “+”, respectively.
According to the rRNA gene content, all rRNA genes had 52.75% A + T, which indicated a trend toward A + C, as observed in other Siluriformes [22,24]. The AT and GC skews were positive (0.2369) and negative (−0.1463), respectively (Table 2). The A. maculatus 12S rRNA subunit gene was 958 bp, and its 16S rRNA subunit gene was 1675 bp, respectively. As in other fish [4], the two genes were located between tRNAPhe and tRNALeu, and separated by the tRNAVal gene (Figure 1, Table 1). The rRNA gene content of A. maculatus was similar to that of other Siluriformes [41].
3.4. The Control Region
In A. maculatus, the D-loop region was 1075 bp in length, which was longer than in the majority of Siluriformes and was only shorter than that in A. seemanni. A + T content was 62.55%, which is similar to A + T content in other Siluriformes species (Table 2). This was consistent with those of previous reports of other teleosts [45]. Additionally, both AT and GC skews were strongly negative (Table 2).
3.5. Overlapping and Intergenic Spacer Regions
Nine gene boundaries overlapped between adjacent genes, ranging in size from 1 to 10 bp. There was a 10 bp overlap between ATP8 and ATP6 (Table 1), which was observed in several other Siluriformes mtDNA sequences. In addition, 12 intergenic spacers, ranging in size from 1 to 31 bases, contained 66 nucleotides. tRNAAsn and tRNCys constitute the longest intergenic spacer regions (31 bp) (Table 1), which was identified with the results in Clarias fuscus [41].
3.6. Synonymous and Nonsynonymous Substitutions
In general, Ka/Ks ratios reflect evolutionary relationships between homogenous and heterogeneous species and selective pressure at the molecular level [53,54]. A ratio of Ka/Ks > 1, Ka/Ks = 1, and Ka/Ks > 1 indicate that there has been positive selection, neutral mutation, and negative selection, respectively [55]. Four Ariidae mtDNAs (A. maculatus, A. arius, N. thalassina, and O. platypogon) were investigated for their evolutionary rate differences, and the Ka and Ks substitution rates were used to calculate sequence divergences. In all 13 PCGs of the four Ariidae, the average Ka/Ks was 0.1747 and varied from 0.015 (COXII between OP/AM, OP/AA, or OP/NT) to 1.672 (ND4 between AM/NT) (Figure 6). The Ka/Ks ratios indicated that there had been strong purifying selections on multiple genes. In other words, this result showed that natural selection occurred against deleterious mutations with negative selective coefficients [56]. A high percentage of AM/NT and AA/NT variable sites was observed in ND4 (1.672) and ND2 (1.190) among the groups, respectively, whereas the percentage in the COXII gene were the lowest, indicating that ND4 and ND2 underwent positive selection and COXII was the most selectively pressured mitochondrial protein. Furthermore, compared with other species, the ratio of Ka/Ks in A. maculatus and A. arius or N. thalassina was the lowest in all 13 protein-coding genes, implying that these three Ariidae fish had a closer phylogenetic relationship with each other than with O. platypogon, which is consistent with their traditional taxonomy. There were likely differences in selection pressures between the genes, and consequently, they evolved differently. It is interesting to note that the ND2 and ND4 genes have the highest ratios, indicating strand-independent selection pressures.
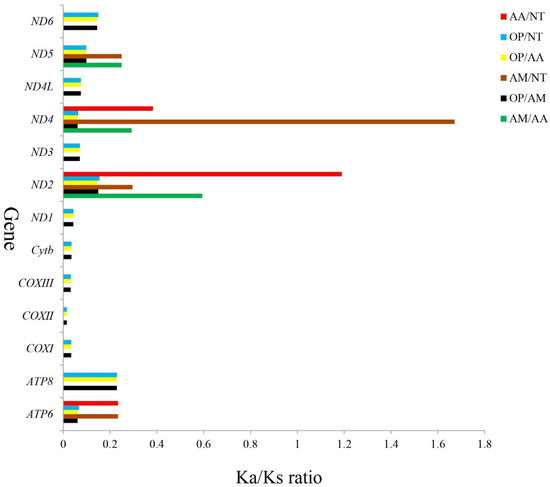
Figure 6.
Ka/Ks ratios for the 13 PCGs among the reference A. maculatus (AM), A. arius (AA), N. thalassina (NT), and O. platypogon (OP).
3.7. Phylogeny
Two methods (BI and ML) were used to establish phylogenetic relationships between 21 species, including the concatenated nucleic acid (Figure 7A) and amino acid (Figure 7B) sequences of the 13 PCGs. Phylogenetic tree topologies of the two superfamilies (Ariidae, Pangasiidae, Bagridae, Mochokidae, Cl ariidae, Siluridae, and Sisoridae) were similar, and strong statistics supported the following relationship among them (Figure 7). The clustering pattern of seven superfamilies was obviously consistent with previous studies [21,22,23,24]. (According to the ML method, 15 closely related genera were identified within the seven superfamilies, and A. maculatus (Arius) was most closely related to A. arius (Arius), N. thalassina (Netuma), O. platypogon (Occidentarius), and B. panamensis (Bagre), which was consistent with a recent study’s findings about nucleotide sequence identity [22,23]. Netuma was most closely related to Arius. To determine the location of Ariidae within Siluriformes, further taxon sampling was required within Ariidae and related superfamilies.
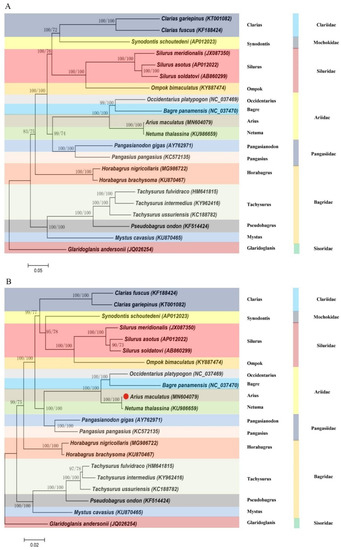
Figure 7.
Phylogenetic trees of A. maculatus relationships from the (A) nucleotide and (B) amino acid datasets. Sequences alignments of mtDNA were analyzed using the RAxML and MrBayes software with the ML and BI method, respectively. Numbers at the nodes are bootstrap values (right) and Bayesian posterior probabilities (left). The accession numbers of the sequences used in the phylogenetic analysis are listed in Table S1.
4. Conclusions
In conclusion, the present study represents common and characteristic features of A. maculatus and other 26 Siluriformes mtDNA, and reveals their phylogenetic relationship. The phylogenetic results strongly support the close relationship between Arius, Netuma, Occidentarius, and Bagre. Our results will provide insight into the basics of evolutionary biology, molecular identification, and conservation of the diverse Siluriformes species, as well as the gene rearrangement process and matrilineal inheritance of A. maculatus.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/genes13112128/s1, Supplementary Figure S1: Graphical illustration showing the AT- and GC-skew in the PCGs of the mtDNA of A. maculatus; Supplementary Table S1: Primers used to verify the accuracy of the assembled mtDNA sequence; Supplementary Table S2: The information of Superfamily, Genera, Species, Size, Genbank number, and Identity in the Siluriformes.
Author Contributions
X.L. and C.L. implemented the molecular genetic studies; M.Y. participated in data analyses and contributed to prepare the figures and tables; M.Y. and Z.Y. conducted data analyses and drafted the manuscript; K.Z. directed this study and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Laboratory of Lingnan Modern Agriculture Project (NT2021008); Financial Fund of Ministry of Agriculture and Rural afairs of China (NHYYSWZZZYKZX2020).
Institutional Review Board Statement
All fsh sampling in this study was permitted by the Animal Care and Use Committee of South China Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences (No. SCSFRI96-253), and conformed with the regulations and guidelines established by this committee.
Informed Consent Statement
This study did not involve humans.
Data Availability Statement
The generated mitochondrial DNA has been submitted to the GenBank database under the accession number MN604079. Moreover, the experimental data involved in this article can be obtained by the corresponding author.
Acknowledgments
We would like to thank the support of the Young Science and Technology Talents Training Fund of South China Agricultural University.
Conflicts of Interest
The authors report no conflict of interest.
References
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [CrossRef] [PubMed]
- Lei, R.; Shore, G.D.; Brenneman, R.A.; Engberg, S.E.; Sitzmann, B.D.; Bailey, C.A.; Kimmel, L.M.; Randriamampionona, R.; Ranaivoarisoa, J.F.; Louis, E.J. Complete sequence and gene organization of the mitochondrial genome for Hubbard′s sportive lemur (Lepilemur hubbardorum). Gene 2010, 464, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.C.; Gong, F.H.; Liu, T.T.; Guo, H.Y.; Zhang, N.; Zhu, K.C.; Jiang, S.G. Shotgun assembly of the mitochondrial genome from Fenneropenaeus penicillatus with phylogenetic consideration. Mar. Genom. 2015, 24, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, J.; Li, C.; Morris, M.; Conn, J.E.; Lima José, B.; Povoa, M.M.; Wilkerson, R.C. Analysis of the evolutionary forces shaping mitochondrial genomes of a Neotropical malaria vector complex. Mol. Phylogenet. Evol. 2011, 58, 469–477. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhu, K.C.; Liang, Y.Y.; Wu, N.; Guo, H.Y.; Zhang, N.; Jiang, S.G.; Zhang, D.C. Sequencing and characterization of the complete mitochondrial genome of Japanese Swellshark (Cephalloscyllium umbratile). Sci. Rep. 2017, 7, 15299. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Ma, C.; Li, X.; Xu, Z.; Feng, N.; Ma, L. The complete mitochondrial genome sequence and gene organization of the mud crab (Scylla paramamosain) with phylogenetic consideration. Gene 2013, 519, 120–127. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef]
- Chen, H.; Lin, L.L.; Chen, X.; Ai, W.M.; Chen, S.B. Complete mitochondrial genome and the phylogenetic position of the Blotchy swell shark Cephaloscyllium umbratile. Mitochondrial DNA A 2016, 27, 3045–3047. [Google Scholar] [CrossRef]
- Gissi, C.; Iannelli, F.; Pesole, G. Evolution of the mitochondrial genome of metazoa as exemplified by comparison of congeneric species. Heredity 2008, 101, 301–320. [Google Scholar] [CrossRef]
- Oliveira, M.T.; Barau, J.G.; Martins-Junqueira, A.C.; Feijao, P.C.; da Rosa, A.C.; Abreu, C.F.; Azeredo-Espin, A.M.; Lessinger, A.C. Structure and evolution of the mitochondrial genomes of Haematobia irritans and Stomoxis calcitrans: The Muscidae (Diptera: Calyptratae) perspective. Mol. Phylogenet. Evol. 2008, 48, 850–857. [Google Scholar] [CrossRef]
- Behura, S.K.; Lobo, N.F.; Haas, B.; Debruyn, B.; Lovin, D.D.; Shumway, M.F.; Puiu, D.; Romero-Severson, J.; Nene, V.; Severson, D.W. Complete sequences of mitochondria genomes of Aedes aegypti and Culex quinquefasciatus and comparative analysis of mitochondrial DNA fragments inserted in the nuclear genomes. Insect. Biochem. Mol. Biol. 2011, 41, 770–777. [Google Scholar] [CrossRef]
- Cameron, S.L. How to sequence and annotate insect mitochondrial genomes for systematic and comparative genomics research. Syst. Entomol. 2014, 39, 400–411. [Google Scholar] [CrossRef]
- Stepien, C.A.; Kocher, T.D. Molecules and morphology in studies of fish evolution. In Molecular Systematics of Fishes; Elsevier: New York, NY, USA, 1997; pp. 1–11. [Google Scholar]
- Miya, M.; Nishida, M. Use of mitogenomic information in teleostean molecular phylogenetics: A tree-based exploration under the maximumparsimony optimality criterion. Mol. Phylogenet. Evol. 2000, 17, 437–455. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.S.; Hou, Y.Y.; Ueng, Y.T.; Wang, J.P.; Chen, H.C. Estimates of age, growth and mortality of spotted catfish, Arius maculatus (Thunberg, 1792), off the Coast of Yunlin, Southwestern Taiwan. Afr. J. Biotechnol. 2011, 10, 15416–15421. [Google Scholar] [CrossRef]
- Maitra, S.; Harikrishnan, M.; Nidhin, B. Feeding strategy, dietary overlap and resource partitioning among four mesopredatory catfishes of a tropical estuary. J. Fish Biol. 2019, 96, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, C.J. Checklist of catfishes, recent and fossil (Osteichthyes: Siluriformes), and catalogue of siluriform primary types. Zootaxa 2007, 1418, 1–628. [Google Scholar] [CrossRef]
- Sczepanski, T.S.; Noleto, R.B.; Cestari, M.M.; Artoni, R.F. A comparative study of two marine catfish (Siluriformes, Ariidae): Cytogenetic tools for determining cytotaxonomy and karyotype evolution. Micron 2010, 41, 193–197. [Google Scholar] [CrossRef]
- Betancur, R.R.; Arturo, A.P.; Bermingham, E.; Cooke, R. Systematics and biogeography of New World sea catfishes (Siluriformes: Ariidae) as inferred from mitochondrial, nuclear, and morphological evidence. Mol. Phylogenet. Evol. 2007, 45, 339–357. [Google Scholar] [CrossRef]
- Betancur, R.R. Molecular phylogenetics and evolutionary history of ariid catfishes revisited: A comprehensive sampling. BMC Evol Biol. 2009, 9, 175. [Google Scholar] [CrossRef]
- Covain, R.; Fisch-Muller, S.; Oliveira, C.; Mol, J.H.; Montoya-Burgos, J.I.; Dray, S. Molecular phylogeny of the highly diversiied catish subfamily Loricariinae (Siluriformes 5421, Loricariidae) reveals incongruences with morphological classification. Mol. Phylogenet. Evol. 2016, 94, 492–517. [Google Scholar] [CrossRef]
- Llera-Herrera, R.; Ramirez-Perez, J.S.; Saavedra-Sotelo, N.C. Complete mitochondrial genome of cominate sea catfish Occidentarius platypogon (Siluriformes: Ariidae). Mitochondrial DNA B 2017, 2, 337–338. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Perez, J.S.; Saavedra-Sotelo, N.C.; Llera-Herrera, R.; Abadia-Chanona, Q.Y. Complete mitochondrial genome of the Chihuil sea catfish Bagre panamensis (Siluriformes: Ariidae). Mitochondrial DNA B 2017, 2, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.F.; Ou, Y.J.; Wen, J.F.; Li, J.E. The complete mitochondrial genome of Arius arius (Siluriformes: Ariidae). Mitochondrial DNA B 2016, 1, 551–552. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tang, M.; Hardman, C.J.; Ji, Y.; Meng, G.; Liu, S.; Tan, M.; Yang, S.; Moss, E.D.; Wang, J.; Yang, C.; et al. High-throughput monitoring of wild bee diversity and abundance via mitogenomics. Methods Ecol. Evol. 2015, 6, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, W.; Fukunaga, T.; Isagozawa, R.; Nishida, M.W. MitoFish and MitoAnnotator: A mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol. Biol. Evol. 2013, 30, 2531–2540. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2007, 33, 686–689. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, 575–581. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Rozas, J. DNA sequence polymorphism analysis using DnaSP. In Bioinformatics for DNA Sequence Analysis; Methods in Molecular Biology Series; Posada, D., Ed.; Humana Press: Totowa, NJ, USA, 2009; Volume 537, pp. 337–350. [Google Scholar]
- Junqueira, A.C.M.; Lessinger, A.C.; Torres, T.T.; da Silva, F.R.; Vettore, A.L.; Arruda, P.; Espin, A.M.L.A. The mitochondrial genome of the blowfly Chrysomya chloropyga (Diptera: Calliphoridae). Gene 2004, 339, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. jModelTest: Phylogenetic Model Averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Diego, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Nakatani, M.; Miya, M.; Mabuchi, K.; Saitoh, K.; Nishida, M. Evolutionary history of Otophysi (Teleostei), a major clade of the modern freshwater fishes: Pangaean origin and Mesozoic radiation. BMC Evol. Biol. 2011, 11, 177. [Google Scholar] [CrossRef]
- Wang, J.L.; Shen, T.; Ju, J.F.; Yang, G.A. The complete mitochondrial genome of the Chinese longsnout catfish Leiocassis longirostris (Siluriformes: Bagridae) and a time-calibrated phylogeny of ostariophysan fishes. Mol. Biol. Rep. 2011, 38, 2507–2516. [Google Scholar] [CrossRef]
- Zhou, C.J.; Wang, X.Z.; Guan, L.H.; He, S.P. The complete mitochondrial genome of Clarias fuscus (Teleostei, Siluriformes: Clariidae). Mitochondrial DNA 2015, 26, 270–271. [Google Scholar] [CrossRef]
- Han, C.; Li, Q.; Xu, J.Q.; Li, X.F.; Huang, J.R. Characterization of Clarias gariepinus mitochondrial genome sequence and a comparative analysis with other catfishes. Biologia 2015, 70, 1245–1253. [Google Scholar] [CrossRef]
- Yang, W.Z.; Zhang, Y.; Feng, S.Q.; Liu, L.J.; Li, Z.H. The first complete mitochondrial genome of the Japanese beetle Popillia japonica (Coleoptera: Scarabaeidae) and its phylogenetic implications for the superfamily Scarabaeoidea. Int. J Biol. Macromol. 2018, 118, 1406–1413. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Zhou, L.; Zhou, X.Y.; Yang, W.T.; Zhang, X.J.; Wang, Y.; Gui, J.F. Unusual AT-skew of Sinorhodeus microlepis mitogenome provides new insights into mitogenome features and phylogenetic implications of bitterling fishes. Int. J Biol. Macromol. 2019, 129, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.M.; Zhu, K.H.; Jiang, H.; Lu, X.T.; Liu, B.J.; Ye, Y.Y.; Jiang, L.H.; Liu, L.Q.; Gong, L. Complete mitochondrial genome of Ophichthus brevicaudatus reveals novel gene order and phylogenetic relationships of Anguilliformes. Int. J. Biol. Macromol. 2019, 135, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.B.; He, Y.F.; Yang, D.G. The complete mitochondrial genome of the Pseudecheneis immaculatus (Siluriformes:Sisoridae). Mitochondrial DNA B 2019, 4, 3120–3121. [Google Scholar] [CrossRef]
- Alam, M.J.; Andriyono, S.; Lee, S.R.; Hossain, M.A.R.; Eunus, A.T.M.; Hassan, M.T.; Kim, H.W. Characterization of the complete mitochondrial genome of Gangetic ailia, Ailia coila (Siluriformes: Ailiidae). Mitochondrial DNA B 2019, 4, 2258–2259. [Google Scholar] [CrossRef]
- Barman, A.S.; Singh, M.; Pandey, P.K. Complete mitochondrial genome of near threatened butter Catfish Ompok bimaculatus (Siluriformes: Siluridae). Mitochondrial DNA B 2017, 2, 313–314. [Google Scholar] [CrossRef][Green Version]
- Zhou, C.L.; Tian, J.; Yang, C.G. The complete mitochondrial genome sequence of Hemibagrus sp. (Siluriformes: Bagridae), molecular data for species identification. Mitochondrial DNA A 2016, 27, 1914–1915. [Google Scholar] [CrossRef]
- Lian, Z.Q.; Wu, X.D.; Xiao, W.; Sai, Q.Y.; Gun, S.B. Complete sequence and characterization of the Silurus lanzhouensis (Siluriformes: Siluridae) mitochondrial genome. Mitochondrial DNA A 2016, 27, 2483–2484. [Google Scholar] [CrossRef]
- Kim, N.K.; Gietbong, F.Z.; Andriyono, S.; Kim, A.R.; Kim, H.W. The complete mitogenome of Bagrid catfish Chrysichthys nigrodigitatus (Siluriformes: Claroteidae). Mitochondrial DNA B 2018, 3, 1239–1240. [Google Scholar] [CrossRef]
- Janke, A.; Pääbo, S. Editing of a tRNA anticodon in marsupial mitochondria changes its codon recognition. Nucleic. Acids. Res. 1993, 21, 1523–1525. [Google Scholar] [CrossRef]
- Shen, X.; Ren, J.; Cui, Z.; Sha, Z.; Wang, B.; Xiang, J.; Liu, B. The complete mitochondrial genomes of two common shrimps (Litopenaeus vannamei and Fenneropenaeus chinensis) and their phylogenomic considerations. Gene 2007, 403, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Huang, Y.; Lei, F.M. Comparative mitochondrial genomics and phylogenetic relationships of the Crossoptilon species (Phasianidae, Galliformes). BMC Genom. 2015, 16, 42. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Yang, Z.; Bielawski, J.P. Statistical methods for detecting molecular adaptation. Trends Ecol Evol. 2000, 15, 496–503. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).