
Strategies for Bottlenecks of rAAV-Mediated Expression in Skeletal and Cardiac Muscle of Duchenne Muscular Dystrophy

Abstract
1. Introduction
2. Current Situation of DMD Treatment
3. Clinical Development of Systemic AAV-Mediated Mini/Micro-Dystrophin Gene Therapy
3.1. Sarepta-SRP-9001
3.2. Pfizer-PF-06939926
3.3. Solid Biosciences-SGT-001
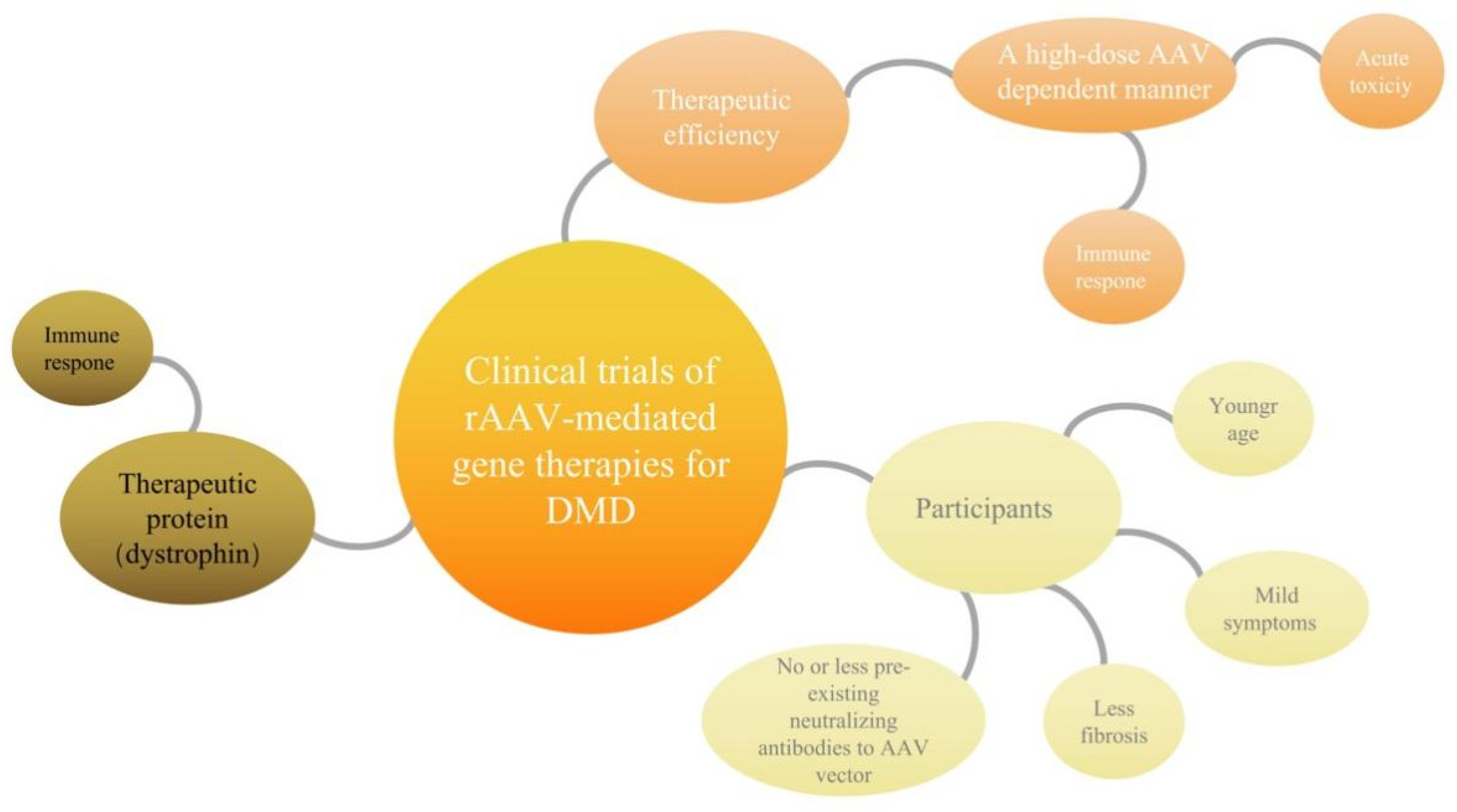
4. Possible Bottlenecks of Clinical Trials of rAAV- Mediated Gene Therapies for DMD
4.1. Immune Response to High-Dose Systemic AAV May Account for Acute Toxicity
4.2. The Mini/Micro-Dystrophin Was Recognized as “Nonconformist” and Destroyed by Overactive Immune Cells, Reducing the Therapeutic Effect
4.3. Fibrosis of Skeletal Muscle Cells and Myocardial Cells Directly Affects the Therapeutic Microenvironment
5. Several Strategies Are Currently under Development to Overcome the Bottlenecks
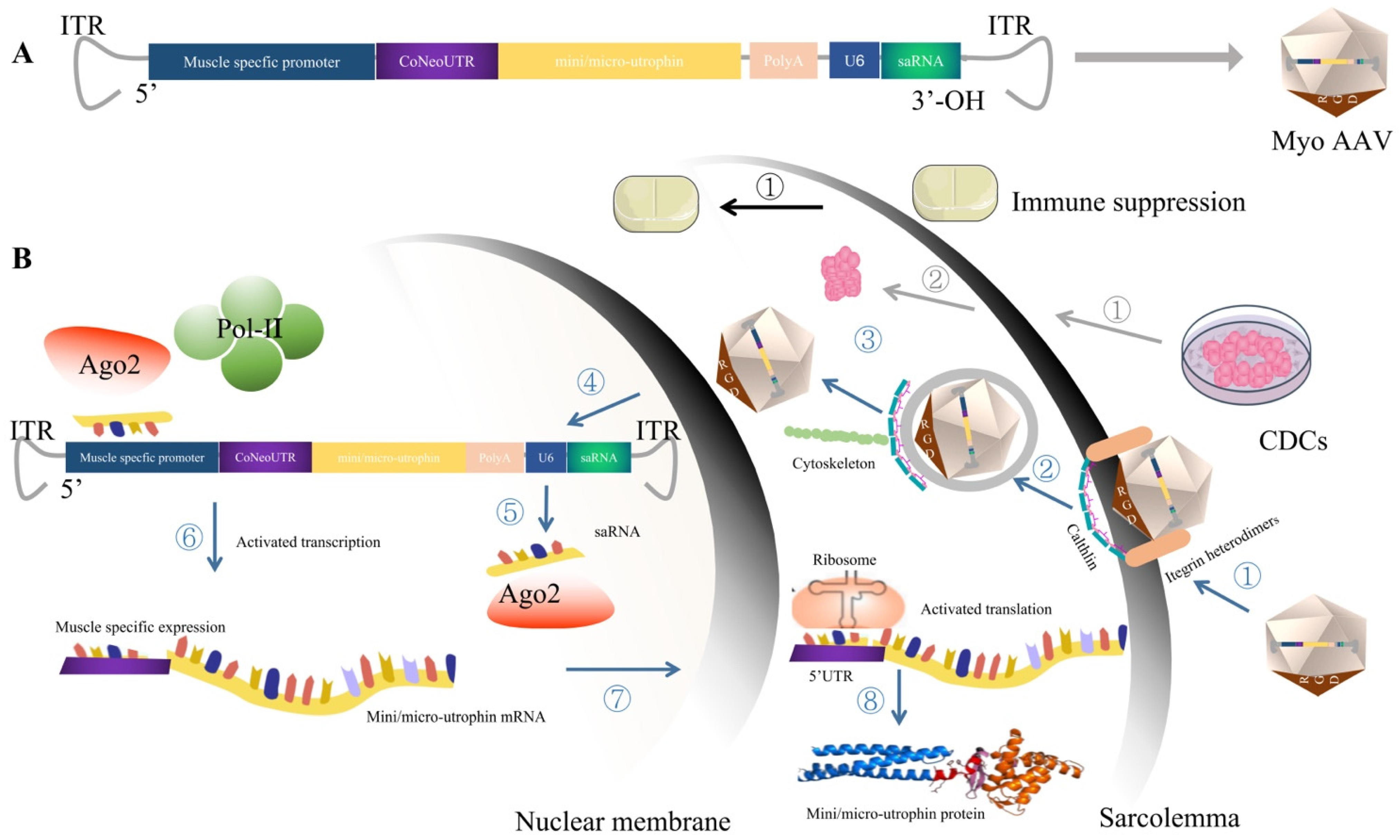
5.1. MyoAAV, Which Targets Striated Muscle Transduction, Presents Great Therapeutic Potential for DMD after Injection of a Low Dose of Virus
5.2. Expression Cassette Structure Optimization
5.2.1. Utrophin as the Expression Cassette to Circumvent the Potential Immune Response of Dystrophin
5.2.2. Small Activation RNA Targeting the Promoter Region of Mini/Micro-Utrophin Can Be Used to Improve the Transcription Level of Mini/Micro-Utrophin
5.2.3. The 5′UTR Region Was Optimized to Enhance the Translation Level of Mini/Micro-Utrophin
5.3. CDCs Therapy Modulates Immune Response and Anti-Fibrosis, Providing an Effective Therapeutic Microenvironment for Gene Therapy of DMD
5.4. Immune Suppression May Be the Promising Approach to Manage Capsid or, Potentially, Transgene Immunogenicity
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moser, H. Duchenne muscular dystrophy: Pathogenetic aspects and genetic prevention. Hum. Genet. 1984, 66, 17–40. [Google Scholar] [CrossRef] [PubMed]
- Birnkrant, D.J.; Panitch, H.B.; Benditt, J.O.; Boitano, L.J.; Carter, E.R.; Cwik, V.A.; Finder, J.D.; Iannaccone, S.T.; Jacobson, L.E.; Kohn, G.L.; et al. American College of Chest Physicians consensus statement on the respiratory and related management of patients with Duchenne muscular dystrophy undergoing anesthesia or sedation. Chest 2007, 132, 1977–1986. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Ibraghimov-Beskrovnaya, O.; Ervasti, J.M.; Leveille, C.J.; Slaughter, C.A.; Sernett, S.W.; Campbell, K.P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 1992, 355, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Henricson, E.K.; Abresch, R.T.; Han, J.J.; Escolar, D.M.; Florence, J.M.; Duong, T.; Arrieta, A.; Clemens, P.R.; Hoffman, E.P.; et al. The cooperative international neuromuscular research group Duchenne natural history study—A longitudinal investigation in the era of glucocorticoid therapy: Design of protocol and the methods used. Muscle Nerve 2013, 48, 32–54. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.P. Three muscular dystrophies: Loss of cytoskeleton-extracellular matrix linkage. Cell 1995, 80, 675–679. [Google Scholar] [CrossRef]
- Matthews, E.; Brassington, R.; Kuntzer, T.; Jichi, F.; Manzur, A.Y. Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2016. [Google Scholar] [CrossRef]
- Cohn, R.D.; van Erp, C.; Habashi, J.P.; Soleimani, A.A.; Klein, E.C.; Lisi, M.T.; Gamradt, M.; ap Rhys, C.M.; Holm, T.M.; Loeys, B.L.; et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat. Med. 2007, 13, 204–210. [Google Scholar] [CrossRef]
- Duboc, D.; Meune, C.; Lerebours, G.; Devaux, J.-Y.; Vaksmann, G.; Bécane, H.-M. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J. Am. Coll Cardiol. 2005, 45, 855–857. [Google Scholar] [CrossRef]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Casimersen: First Approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic. Acid Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Dowling, J.J. Eteplirsen therapy for Duchenne muscular dystrophy: Skipping to the front of the line. Nat. Rev. Neurol. 2016, 12, 675–676. [Google Scholar] [CrossRef]
- Frank, D.E.; Schnell, F.J.; Akana, C.; El-Husayni, S.H.; Desjardins, C.A.; Morgan, J.; Charleston, J.S.; Sardone, V.; Domingos, J.; Dickson, G.; et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology 2020, 94, e2270–e2282. [Google Scholar] [CrossRef]
- Dhillon, S. Viltolarsen: First Approval. Drugs 2020, 80, 1027–1031. [Google Scholar] [CrossRef]
- Godfrey, C.; Muses, S.; McClorey, G.; Wells, K.E.; Coursindel, T.; Terry, R.L.; Betts, C.; Hammond, S.; O’Donovan, L.; Hildyard, J.; et al. How much dystrophin is enough: The physiological consequences of different levels of dystrophin in the mdx mouse. Hum. Mol. Genet. 2015, 24, 4225–4237. [Google Scholar] [CrossRef]
- Gushchina, L.V.; Frair, E.C.; Rohan, N.; Bradley, A.J.; Simmons, T.R.; Chavan, H.D.; Chou, H.-J.; Eggers, M.; Waldrop, M.A.; Wein, N.; et al. Lack of Toxicity in Nonhuman Primates Receiving Clinically Relevant Doses of an AAV9.U7snRNA Vector Designed to Induce Exon 2 Skipping. Hum. Gene Ther. 2021, 32, 882–894. [Google Scholar] [CrossRef]
- Wein, N.; Dunn, D.M.; Waldrop, M.A.; Gushchina, L.V.; Frair, E.C.; Weiss, R.B.; Flanigan, K.M. Absence of Significant Off-Target Splicing Variation with a U7snRNA Vector Targeting Exon 2 Duplications. Hum. Gene Ther. 2021, 32, 1346–1359. [Google Scholar] [CrossRef]
- Clinicaltrials.gov. AAV9 U7snRNA Gene Therapy to Treat Boys with DMD Exon 2 Duplications. Available online: https://clinicaltrials.gov/ct2/show/NCT04240314 (accessed on 27 October 2022).
- Maynard, L.H.; Humbert, O.; Peterson, C.W.; Kiem, H.-P. Genome editing in large animal models. Mol. Ther. 2021, 29, 3140–3152. [Google Scholar] [CrossRef]
- Amoasii, L.; Hildyard, J.C.W.; Li, H.; Sanchez-Ortiz, E.; Mireault, A.; Caballero, D.; Harron, R.; Stathopoulou, T.-R.; Massey, C.; Shelton, J.M.; et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 2018, 362, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.-J.J.; Kühner, K.; Kühn, R.; Werfel, S.; Engelhardt, S.; Wurst, W.; Ortiz, O. Development of an intein-mediated split-Cas9 system for gene therapy. Nucleic. Acids Res. 2015, 43, 6450–6458. [Google Scholar] [CrossRef] [PubMed]
- Moretti, A.; Fonteyne, L.; Giesert, F.; Hoppmann, P.; Meier, A.B.; Bozoglu, T.; Baehr, A.; Schneider, C.M.; Sinnecker, D.; Klett, K.; et al. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat. Med. 2020, 26, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L.; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, F.; Köther, K.; Schmidt, K.; Weghofer, M.; Raupp, C.; Nieto, K.; Kuck, A.; Gerlach, B.; Böttcher, B.; Müller, O.J.; et al. The assembly-activating protein promotes capsid assembly of different adeno-associated virus serotypes. J. Virol. 2011, 85, 12686–12697. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug. Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- England, S.B.; Nicholson, L.V.; Johnson, M.A.; Forrest, S.M.; Love, D.R.; Zubrzycka-Gaarn, E.E.; Bulman, D.E.; Harris, J.B.; Davies, K.E. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature 1990, 343, 180–182. [Google Scholar] [CrossRef]
- Wilton-Clark, H.; Yokota, T. Antisense and Gene Therapy Options for Duchenne Muscular Dystrophy Arising from Mutations in the N-Terminal Hotspot. Genes 2022, 13, 257. [Google Scholar] [CrossRef]
- Salva, M.Z.; Himeda, C.L.; Tai, P.W.; Nishiuchi, E.; Gregorevic, P.; Allen, J.M.; Finn, E.E.; Nguyen, Q.G.; Blankinship, M.J.; Meuse, L.; et al. Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol. Ther. 2007, 15, 320–329. [Google Scholar] [CrossRef]
- Mendell, J.R.; Sahenk, Z.; Lehman, K.; Nease, C.; Lowes, L.P.; Miller, N.F.; Iammarino, M.A.; Alfano, L.N.; Nicholl, A.; Al-Zaidy, S.; et al. Assessment of Systemic Delivery of rAAVrh74.MHCK7.micro-dystrophin in Children With Duchenne Muscular Dystrophy: A Nonrandomized Controlled Trial. J. AMA. Neurol. 2020, 77, 1122–1131. [Google Scholar] [CrossRef]
- Sarepta Therapeutics, I. Sarepta Therapeutics’ Investigational Gene Therapy SRP-9001 for Duchenne Muscular Dystrophy Demonstrates Significant Functional Improvements Across Multiple Studies. Available online: https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics-investigational-gene-therapy-srp-9001 (accessed on 12 October 2022).
- Sarepta Therapeutics, I. Sarepta Therapeutics Announces Top-Line Results for Part 1 of Study 102 Evaluating SRP-9001, Its Investigational Gene Therapy for the Treatment of Duchenne Muscular Dystrophy. Available online: https://investorrelations.Sarepta.com/news-releases/news-release-details/Sarepta-therapeutics-announces-top-line-results-part-1-study-102 (accessed on 5 October 2022).
- Clinicaltrials.gov. A Gene Transfer Therapy Study to Evaluate the Safety of and Expression from SRP-9001 in Participants with Duchenne Muscular Dystrophy (DMD). Available online: https://clinicaltrials.gov/ct2/show/NCT04626674 (accessed on 5 October 2022).
- Zaidman, C.P.C.; Mcdonald, C.; Giblin, K.; Collins, L.; Wang, S.; Upadhyay, S.; Lewis, S.; Malhotra, J.; Griffin, D.A. ENDEAVOR: A Gene Delivery Study to Evaluate the Safety of and Expression from SRP-9001 in Duchenne Muscular Dystrophy. Available online: https://investorrelations.sarepta.com/static-files/a674d68e-823c-43a4-b26c-e6bfc6a5a95b (accessed on 7 October 2022).
- Sarepta Therapeutics, I. Sarepta Therapeutics’ SRP-9001 Shows Sustained Functional Improvements in Multiple Studies of Patients with Duchenne. Available online: https://investorrelations.Sarepta.com/news-releases/news-release-details/Sareptatherapeutics-SRP-9001-shows-sustained-functional (accessed on 7 October 2022).
- Clinicaltrials.gov. A Gene Transfer Therapy Study to Evaluate the Safety and Efficacy of SRP-9001 in Participants with Duchenne Muscular Dystrophy (DMD). Available online: https://clinicaltrials.gov/ct2/show/NCT05096221 (accessed on 5 October 2022).
- Clinicaltrials.gov. Pfizer’s New Phase 1b Results of Gene Therapy in Ambulatory Boys with Duchenne Muscular Dystrophy (DMD) Support Advancement into Pivotal Phase 3 Study. Available online: https://investors.pfizer.com/investor-news/press-releasedetails/2020/Pfizers-New-Phase-1b-Results-of-Gene-Therapy-in-Ambulatory-Boys-with-Duchenne-Muscular-DystrophyDMD-Support-Advancement-into-Pivotal-Phase-3-Study/default.aspx (accessed on 27 October 2022).
- Wang, B.; Li, J.; Fu, F.H.; Chen, C.; Zhu, X.; Zhou, L.; Jiang, X.; Xiao, X. Construction and analysis of compact muscle-specific promoters for AAV vectors. Gene Ther. 2008, 15, 1489–1499. [Google Scholar] [CrossRef]
- Safety and Efficacy of pf-06939926 Gene Therapy in Boys with Duchenne Muscular Dystrophy: Update on Data from the Phase 1b Study | Mda Clinical & Scientific Conference 2022. Available online: https://mdaconference.org/index.php/node/1168 (accessed on 9 December 2021).
- Clinicaltrials.gov. A Phase 3 Study to Evaluate the Safety and Efficacy of pf-06939926 for the Treatment of Duchenne Muscular Dystrophy. Available online: https://clinicaltrials.gov/ct2/show/NCT04281485 (accessed on 5 October 2022).
- Biotech, F. Pfizer Tightens DMD Patient Criteria after Serious Adverse Events Crop up in Phase 3 Gene Therapy Trial | Fiercebiotech. Available online: https://www.fiercebiotech.com/biotech/pfizer-tightening-Duchenne-muscular-dystrophy-phase-3-criteriaadverse-event/ (accessed on 5 October 2022).
- Biotech, F. Pfizer Reports Patient Death in Early-Stage Duchenne Gene Therapy Trial, Halts Enrollment. Available online: https://www.fiercebiotech.com/biotech/pfizer-reports-death-patient-Duchenne-trial-halts-enrolment (accessed on 5 October 2022).
- Duan, D. Systemic AAV Micro-dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2018, 26, 2337–2356. [Google Scholar] [CrossRef]
- BioSpace. FDA Slaps Clinical Hold on Solid Bioscience DMD Gene Therapy Program. Available online: https://www.biospace.com/article/fda-slaps-clinical-hold-on-solid-bioscience-DMD-gene-therapy-program/ (accessed on 6 October 2022).
- BioSpace. FDA Slaps Second Clinical Hold on Solid Biosciences’ DMD Gene Therapy Due to Adverse Event. Available online: https://www.biospace.com/article/fda-slaps-second-clinical-hold-on-solid-biosciences-DMD-gene-therapy-due-toadverse-events/ (accessed on 7 October 2022).
- Zaiss, A.K.; Cotter, M.J.; White, L.R.; Clark, S.A.; Wong, N.C.W.; Holers, V.M.; Bartlett, J.S.; Muruve, D.A. Complement is an essential component of the immune response to adeno-associated virus vectors. J. Virol. 2008, 82, 2727–2740. [Google Scholar] [CrossRef]
- Annoussamy, M.; Lilien, C.; Gidaro, T.; Gargaun, E.; Chê, V.; Schara, U.; Gangfuß, A.; D’Amico, A.; Dowling, J.J.; Darras, B.T.; et al. X-linked myotubular myopathy: A prospective international natural history study. Neurology. 2019, 92, e1852–e1867. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.A.; Rodino-Klapac, L.R.; Goodspeed, K.; Gray, S.J.; Kay, C.N.; Boye, S.L.; Boye, S.E.; George, L.A.; Salabarria, S.; et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther. 2021, 29, 464–488. [Google Scholar] [CrossRef]
- Philippidis, A. Novartis Confirms Deaths of Two Patients Treated with Gene Therapy Zolgensma. Hum. Gene Ther. 2022, 33, 842–844. [Google Scholar] [CrossRef]
- Duan, D. Systemic delivery of adeno-associated viral vectors. Curr. Opin. Virol. 2016, 21, 16–25. [Google Scholar] [CrossRef]
- Kornegay, J.N.; Li, J.; Bogan, J.R.; Bogan, D.J.; Chen, C.; Zheng, H.; Wang, B.; Qiao, C.; Howard, J.F.; Xiao, X. Widespread muscle expression of an AAV9 human mini-dystrophin vector after intravenous injection in neonatal dystrophin-deficient dogs. Mol. Ther. 2010, 18, 1501–1508. [Google Scholar] [CrossRef]
- Hordeaux, J.; Wang, Q.; Katz, N.; Buza, E.L.; Bell, P.; Wilson, J.M. The Neurotropic Properties of AAV-PHP.B Are Limited to C57BL/6J Mice. Mol. Ther. 2018, 26, 664–668. [Google Scholar] [CrossRef]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef]
- Shayakhmetov, D.M.; Di Paolo, N.C.; Mossman, K.L. Recognition of virus infection and innate host responses to viral gene therapy vectors. Mol. Ther. 2010, 18, 1422–1429. [Google Scholar] [CrossRef]
- Mingozzi, F.; High, K.A. Immune responses to AAV vectors: Overcoming barriers to successful gene therapy. Blood 2013, 122, 23–36. [Google Scholar] [CrossRef]
- Lochmüller, H.; Petrof, B.J.; Pari, G.; Larochelle, N.; Dodelet, V.; Wang, Q.; Allen, C.; Prescott, S.; Massie, B.; Nalbantoglu, J.; et al. Transient immunosuppression by FK506 permits a sustained high-level dystrophin expression after adenovirus-mediated dystrophin minigene transfer to skeletal muscles of adult dystrophic (mdx) mice. Gene Ther. 1996, 3, 706–716. [Google Scholar]
- Song, Y.; Morales, L.; Malik, A.S.; Mead, A.F.; Greer, C.D.; Mitchell, M.A.; Petrov, M.T.; Su, L.T.; Choi, M.E.; Rosenblum, S.T.; et al. Non-immunogenic utrophin gene therapy for the treatment of muscular dystrophy animal models. Nat. Med. 2019, 25, 1505–1511. [Google Scholar] [CrossRef]
- Zhou, L.; Lu, H. Targeting fibrosis in Duchenne muscular dystrophy. J. Neuropathol Exp. Neurol. 2010, 69, 771–776. [Google Scholar] [CrossRef]
- Desguerre, I.; Mayer, M.; Leturcq, F.; Barbet, J.-P.; Gherardi, R.K.; Christov, C. Endomysial fibrosis in Duchenne muscular dystrophy: A marker of poor outcome associated with macrophage alternative activation. J. Neuropathol. Exp. Neurol. 2009, 68, 762–773. [Google Scholar] [CrossRef]
- Cordier, L.; Hack, A.A.; Scott, M.O.; Barton-Davis, E.R.; Gao, G.; Wilson, J.M.; McNally, E.M.; Sweeney, H.L. Rescue of skeletal muscles of gamma-sarcoglycan-deficient mice with adeno-associated virus-mediated gene transfer. Mol. Ther. 2000, 1, 119–129. [Google Scholar] [CrossRef]
- Weinmann, J.; Weis, S.; Sippel, J.; Tulalamba, W.; Remes, A.; El Andari, J.; Herrmann, A.-K.; Pham, Q.H.; Borowski, C.; Hille, S.; et al. Identification of a myotropic AAV by massively parallel in vivo evaluation of barcoded capsid variants. Nat. Commun. 2020, 11, 5432. [Google Scholar] [CrossRef]
- El Andari, J.; Renaud-Gabardos, E.; Tulalamba, W.; Weinmann, J.; Mangin, L.; Pham, Q.H.; Hille, S.; Bennett, A.; Attebi, E.; Bourges, E.; et al. Semirational bioengineering of AAV vectors with increased potency and specificity for systemic gene therapy of muscle disorders. Sci. Adv. 2022, 8, eabn4704. [Google Scholar] [CrossRef]
- Tabebordbar, M.; Lagerborg, K.A.; Stanton, A.; King, E.M.; Ye, S.; Tellez, L.; Krunnfusz, A.; Tavakoli, S.; Widrick, J.J.; Messemer, K.A.; et al. Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Cell 2021, 184, 4919–4938. [Google Scholar] [CrossRef]
- Love, D.R.; Hill, D.F.; Dickson, G.; Spurr, N.K.; Byth, B.C.; Marsden, R.F.; Walsh, F.S.; Edwards, Y.H.; Davies, K.E. An autosomal transcript in skeletal muscle with homology to dystrophin. Nature 1989, 339, 55–58. [Google Scholar] [CrossRef]
- Khurana, T.S.; Watkins, S.C.; Chafey, P.; Chelly, J.; Tomé, F.M.; Fardeau, M.; Kaplan, J.C.; Kunkel, L.M. Immunolocalization and developmental expression of dystrophin related protein in skeletal muscle. Neuromuscul. Disord. 1991, 1, 185–194. [Google Scholar] [CrossRef]
- Tomé, F.M.; Matsumura, K.; Chevallay, M.; Campbell, K.P.; Fardeau, M. Expression of dystrophin-associated glycoproteins during human fetal muscle development: A preliminary immunocytochemical study. Neuromuscul. Disord. 1994, 4, 343–348. [Google Scholar] [CrossRef]
- Mesnard-Rouiller, L.; Bismuth, J.; Wakkach, A.; Poëa-Guyon, S.; Berrih-Aknin, S. Thymic myoid cells express high levels of muscle genes. J. Neuroimmunol. 2004, 148, 97–105. [Google Scholar] [CrossRef]
- Blake, D.J.; Love, D.R.; Tinsley, J.; Morris, G.E.; Turley, H.; Gatter, K.; Dickson, G.; Edwards, Y.H.; Davies, K.E. Characterization of a 4.8kb transcript from the Duchenne muscular dystrophy locus expressed in Schwannoma cells. Hum. Mol. Genet. 1992, 1, 103–109. [Google Scholar] [CrossRef]
- Sherratt, T.G.; Vulliamy, T.; Strong, P.N. Evolutionary conservation of the dystrophin central rod domain. Biochem. J. 1992, 287 Pt 3, 755–759. [Google Scholar] [CrossRef]
- Tinsley, J.M.; Blake, D.J.; Roche, A.; Fairbrother, U.; Riss, J.; Byth, B.C.; Knight, A.E.; Kendrick-Jones, J.; Suthers, G.K.; Love, D.R. Primary structure of dystrophin-related protein. Nature 1992, 360, 591–593. [Google Scholar] [CrossRef]
- Tinsley, J.M.; Potter, A.C.; Phelps, S.R.; Fisher, R.; Trickett, J.I.; Davies, K.E. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature 1996, 384, 349–353. [Google Scholar] [CrossRef]
- Gilbert, R.; Nalbanoglu, J.; Tinsley, J.M.; Massie, B.; Davies, K.E.; Karpati, G. Efficient utrophin expression following adenovirus gene transfer in dystrophic muscle. Biochem Biophys Res. Commun. 1998, 242, 244–247. [Google Scholar] [CrossRef]
- Odom, G.L.; Gregorevic, P.; Allen, J.M.; Finn, E.; Chamberlain, J.S. Microutrophin delivery through rAAV6 increases lifespan and improves muscle function in dystrophic dystrophin/utrophin-deficient mice. Mol. Ther. 2008, 16, 1539–1545. [Google Scholar] [CrossRef]
- Cerletti, M.; Negri, T.; Cozzi, F.; Colpo, R.; Andreetta, F.; Croci, D.; Davies, K.E.; Cornelio, F.; Pozza, O.; Karpati, G.; et al. Dystrophic phenotype of canine X-linked muscular dystrophy is mitigated by adenovirus-mediated utrophin gene transfer. Gene Ther. 2003, 10, 750–757. [Google Scholar] [CrossRef]
- Stedman, H.H.; Sweeney, H.L.; Shrager, J.B.; Maguire, H.C.; Panettieri, R.A.; Petrof, B.; Narusawa, M.; Leferovich, J.M.; Sladky, J.T.; Kelly, A.M. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 1991, 352, 536–539. [Google Scholar] [CrossRef]
- Li, L.-C.; Okino, S.T.; Zhao, H.; Pookot, D.; Place, R.F.; Urakami, S.; Enokida, H.; Dahiya, R. Small dsRNAs induce transcriptional activation in human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17337–17342. [Google Scholar] [CrossRef]
- Kang, M.R.; Yang, G.; Place, R.F.; Charisse, K.; Epstein-Barash, H.; Manoharan, M.; Li, L.-C. Intravesical delivery of small activating RNA formulated into lipid nanoparticles inhibits orthotopic bladder tumor growth. Cancer Res. 2012, 72, 5069–5079. [Google Scholar] [CrossRef]
- Kingwell, K. Small activating RNAs lead the charge to turn up gene expression. Nat. Rev. Drug Discov. 2021, 20, 573–574. [Google Scholar] [CrossRef]
- Hashimoto, A.; Sarker, D.; Reebye, V.; Jarvis, S.; Sodergren, M.H.; Kossenkov, A.; Sanseviero, E.; Raulf, N.; Vasara, J.; Andrikakou, P.; et al. Upregulation of C/EBPα Inhibits Suppressive Activity of Myeloid Cells and Potentiates Antitumor Response in Mice and Patients with Cancer. Clin. Cancer Res. 2021, 27, 5961–5978. [Google Scholar] [CrossRef]
- Ingolia, N.T. Ribosome profiling: New views of translation, from single codons to genome scale. Nat. Rev. Genet. 2014, 15, 205–213. [Google Scholar] [CrossRef]
- Kozak, M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell 1986, 44, 283–292. [Google Scholar] [CrossRef]
- Kozak, M. Possible role of flanking nucleotides in recognition of the AUG initiator codon by eukaryotic ribosomes. Nucleic. Acids Res. 1981, 9, 5233–5252. [Google Scholar] [CrossRef]
- Cao, J.; Novoa, E.M.; Zhang, Z.; Chen, W.C.W.; Liu, D.; Choi, G.C.G.; Wong, A.S.L.; Wehrspaun, C.; Kellis, M.; Lu, T.K. High-throughput 5’ UTR engineering for enhanced protein production in non-viral gene therapies. Nat. Commun. 2021, 12, 4138. [Google Scholar] [CrossRef]
- Dutton, L.C.; Dudhia, J.; Catchpole, B.; Hodgkiss-Geere, H.; Werling, D.; Connolly, D.J. Cardiosphere-derived cells suppress allogeneic lymphocytes by production of PGE2 acting via the EP4 receptor. Sci. Rep. 2018, 8, 13351. [Google Scholar] [CrossRef]
- Makkar, R.R.; Smith, R.R.; Cheng, K.; Malliaras, K.; Thomson, L.E.; Berman, D.; Czer, L.S.; Marbán, L.; Mendizabal, A.; Johnston, P.V.; et al. Intracoronary cardiosphere-derived cel.lls for heart regeneration after myocardial infarction (CADUCEUS): A prospective, randomised phase 1 trial. Lancet 2012, 379, 895–904. [Google Scholar] [CrossRef]
- Malliaras, K.; Makkar, R.R.; Smith, R.R.; Cheng, K.; Wu, E.; Bonow, R.O.; Marbán, L.; Mendizabal, A.; Cingolani, E.; Johnston, P.V.; et al. Intracoronary cardiosphere-derived cells after myocardial infarction: Evidence of therapeutic regeneration in the final 1-year results of the CADUCEUS trial (CArdiosphere-Derived aUtologous stem CElls to reverse ventricUlar dySfunction). J. Am. Coll. Cardiol. 2014, 63, 110–122. [Google Scholar] [CrossRef]
- McDonald, C.M.; Marbán, E.; Hendrix, S.; Hogan, N.; Ruckdeschel Smith, R.; Eagle, M.; Finkel, R.S.; Tian, C.; Janas, J.; Harmelink, M.M.; et al. Repeated intravenous cardiosphere-derived cell therapy in late-stage Duchenne muscular dystrophy (HOPE-2): A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2022, 399, 1049–1058. [Google Scholar] [CrossRef]
- Aminzadeh, M.A.; Rogers, R.G.; Fournier, M.; Tobin, R.E.; Guan, X.; Childers, M.K.; Andres, A.M.; Taylor, D.J.; Ibrahim, A.; Ding, X.; et al. Exosome-Mediated Benefits of Cell Therapy in Mouse and Human Models of Duchenne Muscular Dystrophy. Stem Cell Rep. 2018, 10, 942–955. [Google Scholar] [CrossRef]
- Rogers, R.G.; Fournier, M.; Sanchez, L.; Ibrahim, A.G.; Aminzadeh, M.A.; Lewis, M.I.; Marbán, E. Disease-modifying bioactivity of intravenous cardiosphere-derived cells and exosomes in mdx mice. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Chu, W.S.; Ng, J. Immunomodulation in Administration of rAAV: Preclinical and Clinical Adjuvant Pharmacotherapies. Front. Immunol 2021, 12, 658038. [Google Scholar] [CrossRef]
- Verdera, H.C.; Kuranda, K.; Mingozzi, F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol. Ther. 2020, 28, 723–746. [Google Scholar] [CrossRef]
- Wilson, J.M.; Flotte, T.R. Moving Forward After Two Deaths in a Gene Therapy Trial of Myotubular Myopathy. Hum. Gene Ther. 2020, 31, 695–696. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Reiss, U.M.; Tuddenham, E.G.D.; Rosales, C.; Chowdary, P.; McIntosh, J.; Della Peruta, M.; Lheriteau, E.; Patel, N.; Raj, D.; et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014, 371, 1994–2004. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sarepta | Pfizer | Therapeutics Solid Biosciences | |
|---|---|---|---|
| Trial name | An open-label, systemic gene delivery study using commercial process material to evaluate the safety of and expression from SRP-9001 in subjects with Duchenne muscular dystrophy (ENDEAVOR) | A phase 1b multicenter, open-label, single ascending dose study to evaluate the safety and tolerability of pf-06939926 in ambulatory and non-ambulatory subjects with Duchenne muscular dystrophy | A randomized, controlled, open-label, single-ascending dose, phase I/II study to investigate the safety and tolerability, and efficacy of intravenous SGT-001 in male adolescents and children with Duchenne muscular dystrophy |
| ClinicalTrials.gov Identififier | NCT04626674 | NCT03362502 | NCT03368742 |
| Study nature | Phase-1b trial | Phase 1b, open-label, trial | Phase 1/2, open-label, trial |
| Drug name | SRP-9001 | PF-06939926 | SGT-001 |
| AAV-serotype | rAAV-rh74 | rAAV9 | rAAV9 |
| Dose | 1 dose (1.33 × 1014 vg/kg) for cohort 1 | 2 doses (1.0 × 1014 vg/kg, 3.0 × 1014 vg/kg) | 2 doses (5.0 × 1013 vg/kg 2.0 × 1014 vg/kg) |
| Patient number | 38 | 22 | 16 (estimated enrollment) |
| Patient average age | 3 years and older | 4 years and older | 4~17 years |
| Disease stage | Ambulatory and non-ambulatory subjects | Ambulatory and non-ambulatory subjects | Ambulatory and non-ambulatory subjects |
| Corticosteroid use | 3 months on stable weekly dose of oral corticosteroids for cohort 1 | Daily glucocorticoids for at least 3 months | Stable daily dose (or equivalent) of oral corticosteroids ≥ 12 wks |
| Dystrophin gene mutation | Any mutation | Any mutation | Any mutation |
| Pre-Nab to AAV | Negative | Negative | Negative |
| Primary outcome | The change in micro-dystrophin expression in DMD patients treated with SRP-9001. | Safety and tolerability | Safety |
| Secondary outcome | Adverse events, vector shedding, and the development of antibodies to AAVrh74. | Micro-dystrophin expression in biopsy | |
| SAE | Increased transaminases that required corticosteroid treatment. Nausea and vomiting that required intravenous treatment (cohort 1). | More than 40% of patients suffered vomiting, nausea, decreased appetite, and pyrexia. | Complement activation, reduced platelet count, liver dysfunction, and acute kidney injury |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, N.; Song, Y. Strategies for Bottlenecks of rAAV-Mediated Expression in Skeletal and Cardiac Muscle of Duchenne Muscular Dystrophy. Genes 2022, 13, 2021. https://doi.org/10.3390/genes13112021
Li N, Song Y. Strategies for Bottlenecks of rAAV-Mediated Expression in Skeletal and Cardiac Muscle of Duchenne Muscular Dystrophy. Genes. 2022; 13(11):2021. https://doi.org/10.3390/genes13112021
Chicago/Turabian StyleLi, Na, and Yafeng Song. 2022. "Strategies for Bottlenecks of rAAV-Mediated Expression in Skeletal and Cardiac Muscle of Duchenne Muscular Dystrophy" Genes 13, no. 11: 2021. https://doi.org/10.3390/genes13112021
APA StyleLi, N., & Song, Y. (2022). Strategies for Bottlenecks of rAAV-Mediated Expression in Skeletal and Cardiac Muscle of Duchenne Muscular Dystrophy. Genes, 13(11), 2021. https://doi.org/10.3390/genes13112021