
Development of Single Nucleotide Polymorphism and Association Analysis with Growth Traits for Black Porgy (Acanthopagrus schlegelii)

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Materials
2.2. Experimental Method
2.2.1. Extraction of Genomic DNA from Black Porgy
2.2.2. Screening of Candidate SNPs
2.2.3. Genotyping Primer Design
2.2.4. PCR Amplification and Extension Reactions
2.2.5. SNP Locus Genotyping
2.2.6. Genetic Polymorphism Analysis
2.2.7. Correlation and Principal Component Analysis of Growth Traits
2.2.8. Correlation and Amino Acid Sequence Analysis
3. Results and Analysis
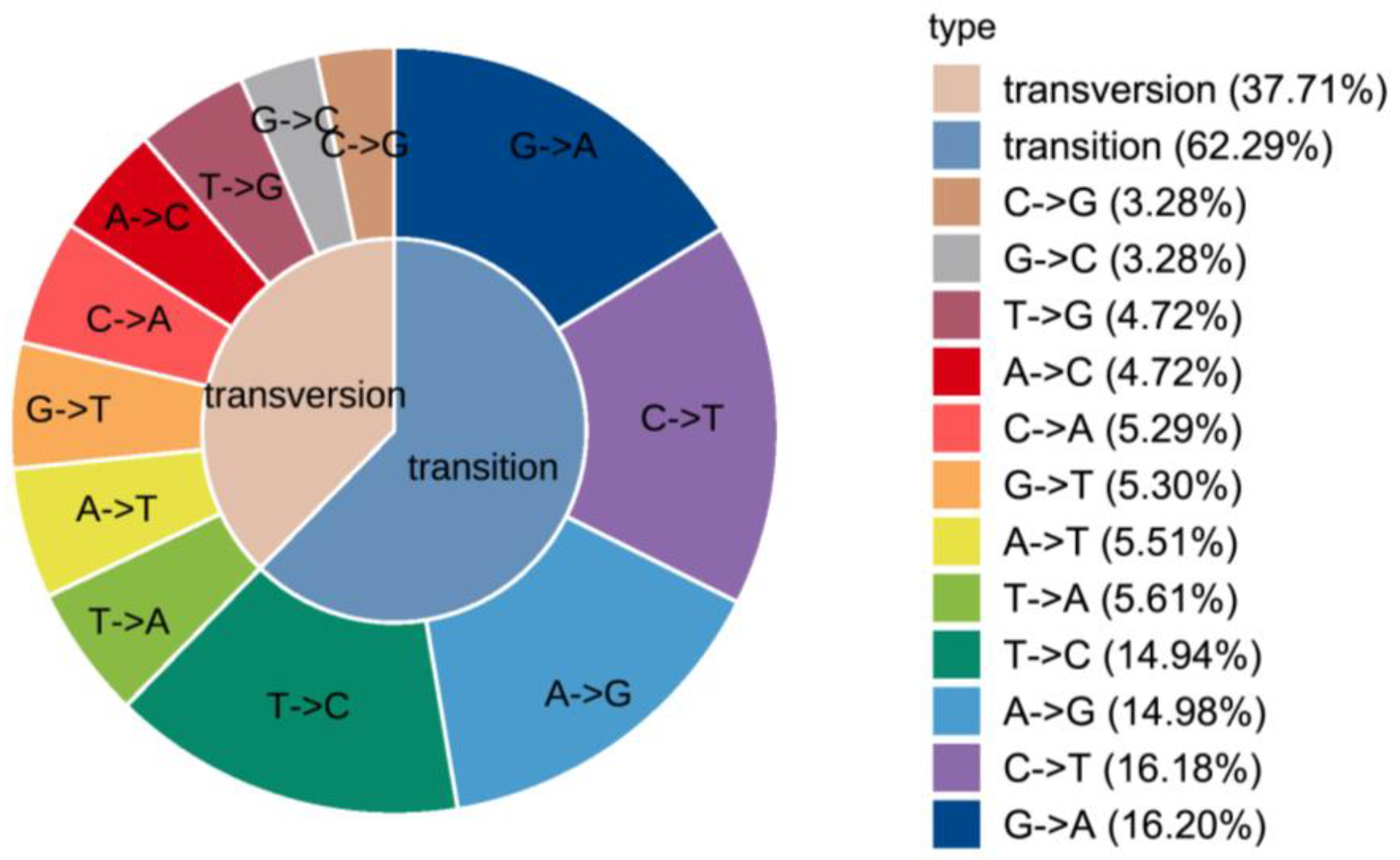
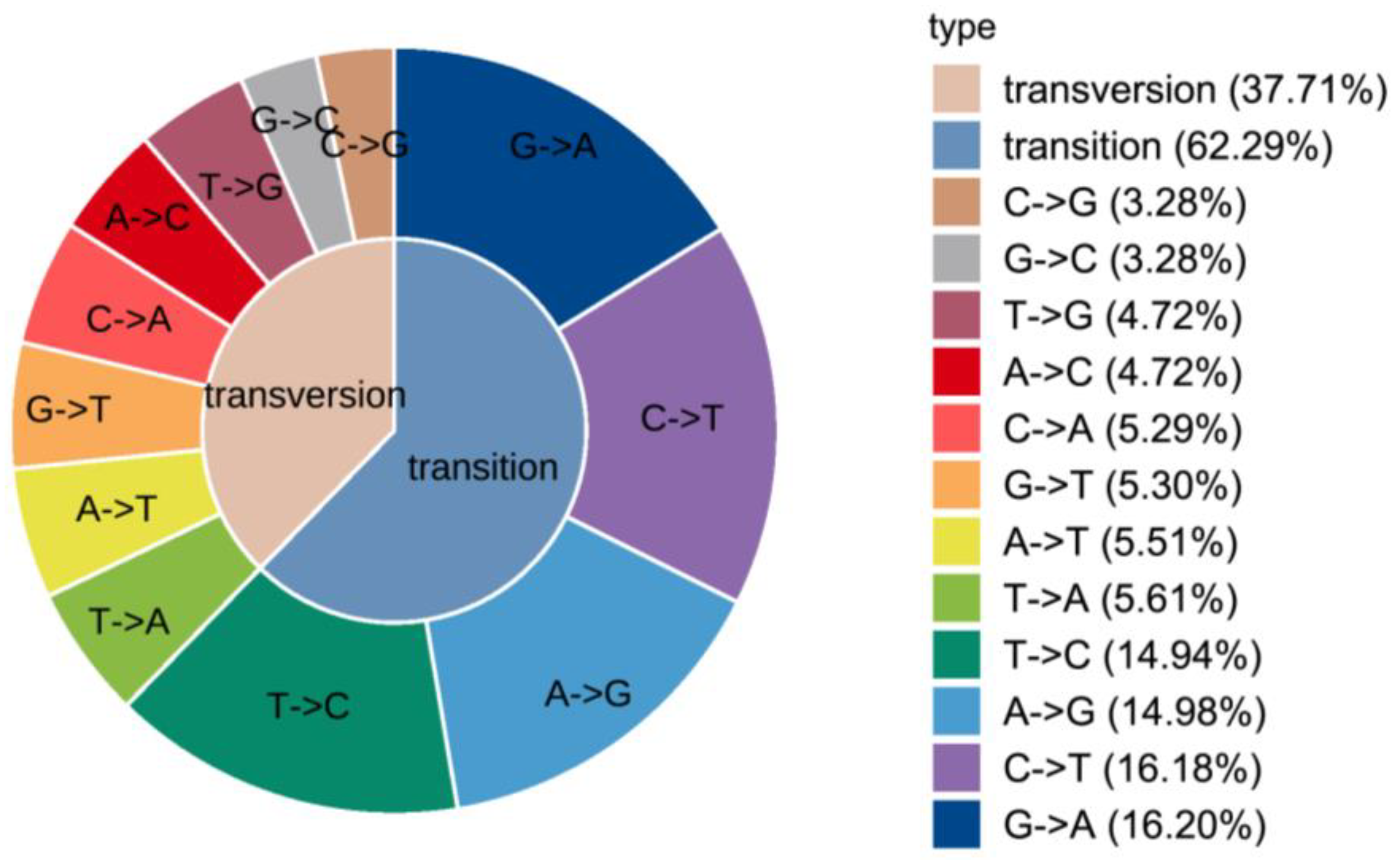
3.1. Transcriptome Candidate SNPs Screening
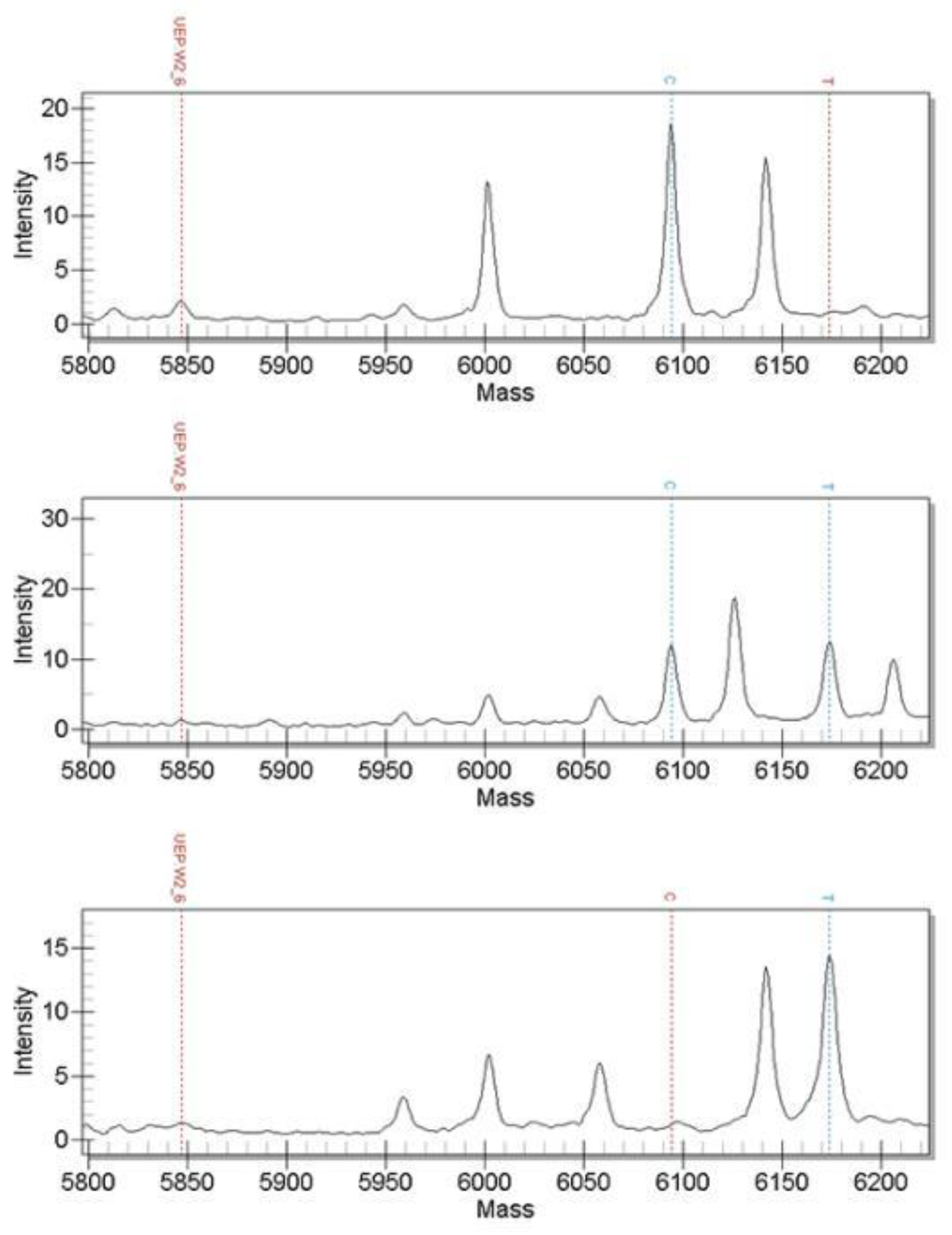
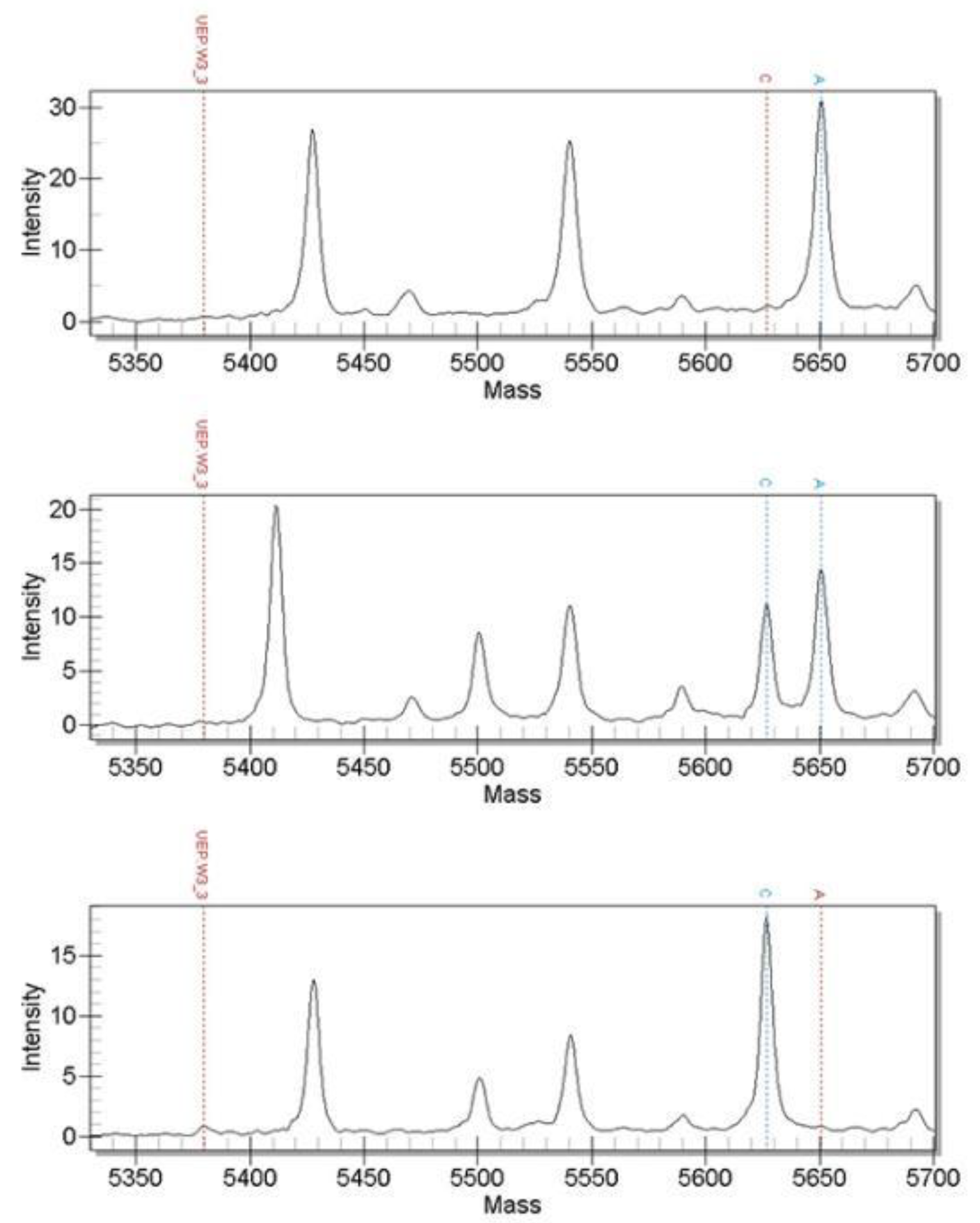
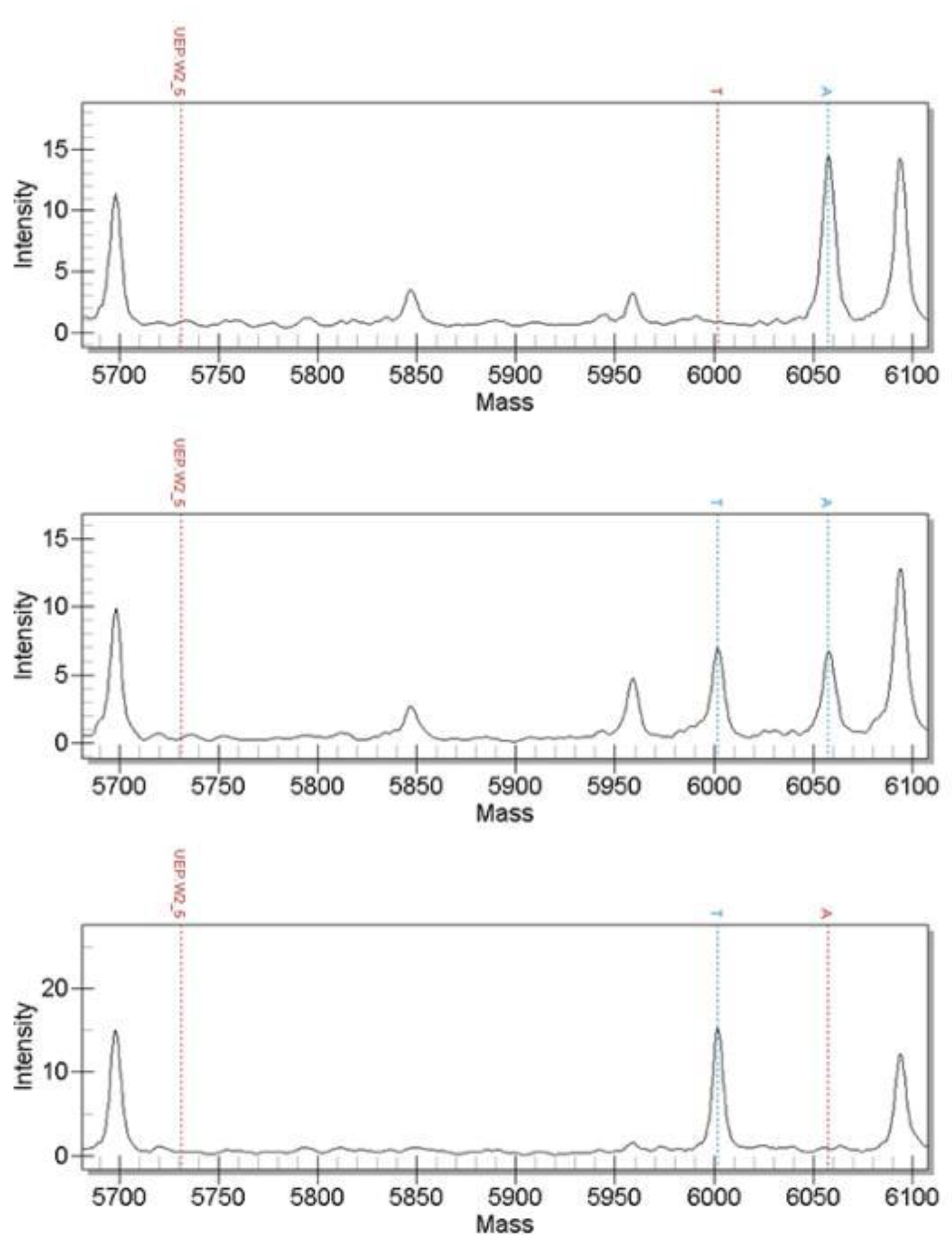
3.2. Time-of-Flight Mass Spectrometry
3.3. Polymorphism and Genetic Parameter Analysis of SNPs
3.4. Correlation and Principal Component Analysis of Growth Traits
3.5. Correlation Analysis of SNPs Markers and Growth Traits
3.6. Amino acid Sequence Analysis
4. Discussion
4.1. Development of SNPs Markers by Transcriptome Sequencing
4.2. Genetic Parameter Analysis of SNPs in the Black Porgy Population
4.3. Principal Component Analysis of Growth Traits of Black Porgy
4.4. Association Analysis of SNPs and Growth Traits of Black Porgy
4.5. The Effect of Base Changes on Amino Acid Sequence
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, R.; Liu, J.; Fan, J.; Zhao, Y. Exploration on the nomenclature and taxonomic status of black porgy. Mar. Sci. 2011, 35, 117–119. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, Z.; Zhu, F.; Jia, C.; Meng, Q.; Cao, G.; Lin, Z.; Zhang, Z. Effects of acute hypothermia on antioxidant enzyme activity and heat shock protein content of black porgy. J. Zool. 2020, 55, 784–792. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, Z.; Liu, H.; Jia, C.; Xu, J.; Zhu, F.; Zhang, Z.; Ren, Z. Histological observation of sex reversal of pond-raised black porgy. Mar. Sci. 2017, 41, 51–58. [Google Scholar]
- Liu, X.; Cui, L.; Li, S.; Jiang, K.; Yuan, X.; Yu, X.; Wang, D.; Wu, F.; Song, D.; Gao, H.; et al. The China Fishery Statistical Yearbook; National Bureau of Statistics of China: Beijing, China, 2022; Volume 2022, p. 22.
- Gao, Z.; Luo, W.; Liu, H.; Zeng, C.; Liu, X.; Yi, S.; Wang, W. Transcriptome analysis and SSR/SNP markers information of the blunt snout bream (Megalobrama amblycephala). PLoS ONE 2012, 7, e42637. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Xie, Y.; Wei, X.; Luo, H.; Chen, D. Development of SSR and SNP molecular markers in Rana spinosa based on transcriptome sequencing. South. Agric. J. 2021, 53, 759–767. [Google Scholar]
- Dai, P.; Huan, P.; Wang, H.X.; Lu, X.; Liu, B.Z. Characterization of a long-chain fatty acid-CoA ligase 1 gene and association between its SNPs and growth traits in the clam Meretrix meretrix. Gene 2015, 566, 194–200. [Google Scholar] [CrossRef]
- Li, G.; Zhao, Y.; Guo, S.; Liu, B.; Chen, Y.; Sun, X.; Feng, J. Dataset on evolution analysis of splenic transcriptome in bighead carp and silver carp. Data Brief 2019, 22, 812–814. [Google Scholar] [CrossRef]
- Rong, M.; Zhang, R.; Tu, J.; Xing, X. Transcriptome differential expression analysis during rapid growth of American shorthaired black mink. J. Anim. Husb. Vet. Med. 2019, 50, 1787–1801. [Google Scholar]
- Zhou, Y.; Fu, H.; Wang, Y.; Huang, H. Genome-wide association study reveals growth-related SNPs and candidate genes in mandarin fish (Siniperca chuatsi). Aquaculture 2022, 550, 737879. [Google Scholar] [CrossRef]
- Lyu, D.; Hu, Y.; Wu, H.; Sun, S.; Wang, W. Estimating breeding values for juvenile body weight using trait associated SNP assisted BLUP in turbot (Scophthalmus maximus). Aquaculture 2019, 508, 46–51. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, J.; Sun, X. Discovery of SNPs related to carp growth based on EST sequences. J. Fish. 2009, 22, 1–7. [Google Scholar]
- You, X.; Shan, X.; Shi, Q. Research advances in the genomics and applications for molecular breeding of aquaculture animals. Aquaculture 2020, 526, 735357. [Google Scholar] [CrossRef]
- Tran, T.T.H.; Nguyen, H.T.; Kim, O.T.P. Characterization of single nucleotide polymorphism in IGF1 and IGF1R genes associated with growth traits in striped catfish (Pangasianodon hypophthalmus Sauvage, 1878). Aquaculture 2021, 538, 736542. [Google Scholar] [CrossRef]
- Ge, C.; Ling, Q.; Li, C.; Zhu, P. Development of growth-related SNP markers for Paramisgurnus dabryanus based on second-generation sequencing. Jiangsu Agric. Sci. 2014, 42, 21–27. [Google Scholar] [CrossRef]
- Li, S.; Liu, H.; Bai, J.; Sun, J. NPY gene polymorphism and its association with growth in largemouth bass. Genom. Appl. Biol. 2018, 37, 4274–4277. [Google Scholar]
- Pan, N.R.; Wang, M.H.; Zhong, L.Q.; Bian, W.J.; Chen, X.H.; Zhang, S.Y. Identification of male-specific SNP markers and development of rapid PCR-based genetic sex identification method in channel catfish (Ictalurus punctatus). Aquaculture 2022, 547, 737535. [Google Scholar] [CrossRef]
- Mao, S.; Bao, S.; Xu, X.; Shen, Y. Screening of SNP markers of mitochondrial ND4L gene in black carp (Mylopharyngodon piceus) and their frequencies in two different body color populations. J. Fish. China 2022, 46, 1–8. [Google Scholar]
- Li, G.; Jin, F.; Zhou, R.; Zhang, W. Genetic diversity analysis of natural populations of Schizothorax wangchiachii based on SNP markers. Acta Hydrobiol. Sin. 2018, 42, 271–276. [Google Scholar]
- Yu, L.; Liu, B.; Liu, J. Screening of discrete SNP markers and analysis of population genetic diversity in Anguilla japonica. Period. Ocean. Univ. China 2018, 48, 37–47. [Google Scholar]
- Zhu, J.; Dai, Q.; Duan, J.; Gao, H. Screening and characteristic analysis of SNP loci in mitochondrial genome of Exopalaemon carinicauda. Fish. Sci. 2021, 40, 387–393. [Google Scholar]
- Zhou, C. Molecular Cloning and Functional Characterization of IGF-1R and Duplicated IGFBP-5 in Grass Carp (Ctenopharyngodon idellus) and SNP Analysis of Different Grass Carp’s Muscle-Growth Control Gene. Master’s Thesis, Shanghai Ocean University, Shanghai, China, 2014. [Google Scholar]
- Lin, Z.; Zhang, Z.; Solberg, M.F.; Zhang, Z. Comparative Transcriptome Analysis of Mixed Tissues of Black Porgy (Acanthopagrus schlegelii) with Differing Growth Rates. Aquac. Res. 2021, 52, 5800–5813. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Alex, X.F.; Garritsen, H.S.P.; Tarhouny, S.E.L.; Zhong, X. A rapid and accurate approach to identify single nucleotide polymorphisms of mitochondrial DNA using MALDI-TOFmass spectrometry. Clin. Chem. Lab. Med. 2008, 46, 299–305. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H. Study of the Matrix for Drug Small Molecules and Their Analysis Application Based on Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry. Master’s Thesis, Southwest University, Chongqing, China, 2021. [Google Scholar]
- Zhao, H.; Wang, W.; Zhang, R.; Zeng, C. Research on high-throughput time-of-flight mass spectrometry genotyping method. Prog. Biochem. Biophys. 2005, 32, 667–672. [Google Scholar]
- Wang, H. SNPs Identification of Growth-Related Genes and Correlation Analysis on Growth Traits in Sinipercid Species. Ph.D. Thesis, Sun Yat-Sen University, Guangzhou, China, 2014. [Google Scholar]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a Genetic Linkage Map in Man Using Restriction Fragment Length Polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar]
- Quan, Y.; Ma, D.; Bai, J.; Liu, H. Screening of largemouth bass transcriptome SNPs and their association with growth. Acta Hydrobiol. Sin. 2016, 40, 1128–1134. [Google Scholar] [CrossRef]
- Wang, P. Development and Application of SNP in Large Yellow Croaker (Larimichthys crocea) Based on Genome and Transcriptome. Master’s Thesis, Jimei University, Xiamen, China, 2016. [Google Scholar]
- Li, S.; Bai, J.; Zhao, L.; Zhu, X. Development of EST-SNP markers for largemouth bass and their correlation with growth traits. Mar. Fish. 2018, 40, 38–46. [Google Scholar]
- Sun, X.; Li, S.; Jiang, P.; Bai, J. Analysis of Growth Traits-related Genes and SNP Markers of Ctenopharyngodon idella by RNA-Seq Technology. J. Fish. China 2021, 45, 333–344. [Google Scholar]
- Liu, H.; Liu, J.; Yang, M.; Wang, Y. SSR and SNP Polymorphic Feature Analysis Based on Cheilinus undulatus Transcriptome. Genom. Appl. Biol. 2020, 39, dsac028. [Google Scholar]
- Yu, Y. Development of molecular markers and their applications in selective breeding of the Pacific white shrimp, Litopeneaus vannamei. Ph.D. Thesis, Graduate School of Chinese Academy of Sciences (Institute of Oceanography), Qingdao, China, 2014. [Google Scholar]
- Luo, H.; Ye, H.; Xiao, S.; Wang, Z. Development of SNP markers associated with immune-related genes of Schizothorax prenanti. Conserv. Genet. Resour. 2016, 8, 223–226. [Google Scholar] [CrossRef]
- Chang, L.; Ren, D.; Jiao, L.; Liu, P. Comparison of MALDI TOF MS and fluorescent quantitation for folate metabolism enzymes SNPs detection. Chin. J. Fam. Plann. 2021, 29, 1411–1413. [Google Scholar]
- Wang, T.; Huang, Z.; Ma, A.; Ma, B. SNP marker development and polymorphism analysis of Scophthalmus maximus based on transcriptome data. Oceanol. ET Limnol. Sin. 2014, 45, 1300–1307. [Google Scholar]
- Lin, X.; Xiao, S.; Li, W.; Wang, Z. Development and validation of sex-specific SNP markers in Larimichthys crocea. J. Fish. China 2018, 42, 1329–1337. [Google Scholar]
- Kim, M.J.; Wang, A.R.; Kim, S.S.; Kim, I. Development and validation of microsatellite markers for the tiny dragonfly, Nannophya pygmaea (Odonata: Libellulidae), which is endangered in South Korea. Appl. Entomol. Zool. 2018, 53, 151–156. [Google Scholar] [CrossRef]
- Vaiman, D.; Mercier, D.; Moazami, G.K.; Guérin, G. A set of 99 cattle microsatellite, characterization, synteny mapping and polymorphism. Mamm. Genome 1994, 5, 288–297. [Google Scholar] [CrossRef]
- Peng, M.; Liang, X.; Fang, R.; Zhu, T. Detection of SNPs and SSR of amylase (AMY) gene in Siniperca chuatsi. J. Jinan Univ. (Nat. Sci.) 2013, 34, 96–100. [Google Scholar]
- Yang, X.; Ma, W.; Yang, J.; Qiu, X. SNPs Screening of B-FABP Gene and Their Association with Growth Traits in Takifugu rubripes. J. Guangdong Ocean Univ. 2021, 41, 28–34. [Google Scholar]
- Yang, Y.; Xiang, M.; Ye, X.; Ye, H. Association Analysis of SNP Markers and Growth Traits in Schizothorax prenanti. J. Fish. Sci. China 2018, 25, 278–285. [Google Scholar]
- Ma, J. Cloning and Expression Determination of Cytochrome p450 3a137 Gene Complementary DNA in Silver Carp. Master’s Thesis, Henan Normal University, Xinxiang, China, 2013. [Google Scholar]
- Kashiwada, S.; Hinton, D.E.; Kullman, S.W. Functional characterization of medaka CYP3A38 and CYP3A40 Kinetics and catalysis by expression in a recombinant baculovirus system. Comp. Biochem. Physiol. 2005, 141, 338–348. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, X.; Wang, C.; Chen, M. Bioaccumulation and the expression of hepatic cytochrome P450 genes in marine medaka (Oryzias melastigma) exposed to difenoconazole. J. Environ. Sci. 2017, 52, 98–104. [Google Scholar] [CrossRef]
- Fu, G.; Yang, X. The correlation of pregnane X receptor and cytochrome P450 3A in grass carp (Ctenopharyngodon idellus). Mar. Fish. 2012, 34, 76–82. [Google Scholar]
- Fu, G.; Yang, X.; Yu, W.; Huang, X. Induced expression of cytochrome P450 3A gene in grass carp (Ctenopharyngodon idellus) kidney cells and analysis of its enzymatic activity. J. Fish. Sci. China 2011, 18, 720–727. [Google Scholar]
- Li, X.; Liu, Y.; Cheng, J.; Liu, P. Identification and expression analysis of a fibrinogen alpha chain-like gene in Atlantic salmon (Salmo salar). Aquac. Rep. 2022, 22, 100919. [Google Scholar] [CrossRef]
- Xie, F.J.; Zhang, Z.P.; Lin, P.; Wang, Y.L. Identification of immune responsible fibrinogen beta chain in the liver of large yellow croaker using a modified annealing control primer system. Fish Shellfish Immunol. 2009, 27, 202–209. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Concentration | Volume (1 rxn/μL) | Supplier |
|---|---|---|---|
| SAP Buffer | 10× | 0.17 | ThermoFisher (Waltham, MA, USA) |
| SAP Enzyme | 1.7 U/μl | 0.3 | ThermoFisher (Waltham, MA, USA) |
| Water | / | 1.53 | Sangon Bio (Shanghai, China) |
| PCR product | / | 5 | |
| Total volume | 7 |
| Component | Concentration | Volume (1 rxn/μL) | Supplier |
|---|---|---|---|
| iPlex Buffer | 10× | 0.2 | SEQUENOM (San Diego, CA, USA) |
| iPlex termination mix | 10× | 0.2 | SEQUENOM (San Diego, CA, USA) |
| iPlex enzyme | 2 U/μl | 0.041 | SEQUENOM (San Diego, CA, USA) |
| SAP Enzyme | 1.7 | 0.3 | ThermoFisher (Waltham, MA, USA) |
| Extension Primer | 10 μm | 0.804 | Sangon Bio (Shanghai, China) |
| Water | / | 0.755 | Sangon Bio (Shanghai, China) |
| Purified PCR product | / | 7 | |
| Total volume | 9 |
| Traits | Body Weight (BW) | Full Length (FL) | Body Length (BL) | Body Height (BH) |
|---|---|---|---|---|
| BW | 1 | |||
| FL | 0.970 ** | 1 | ||
| BL | 0.974 ** | 0.994 ** | 1 | |
| BH | 0.952 * | 0.969 ** | 0.969 ** | 1 |
| Component | Initial Component | Sum of Squares of Extracted Component | ||||
|---|---|---|---|---|---|---|
| Total | Variance Ratio/% | Accumulation Variance Ratio/% | Total | Variance Ratio/% | Accumulation Variance Ratio/% | |
| BW | 2.960 | 73.996 | 73.996 | 2.960 | 73.996 | 73.996 |
| FL | 0.999 | 24.977 | 98.972 | |||
| BL | 0.035 | 0.882 | 99.854 | |||
| BH | 0.006 | 0.146 | 100.000 | |||
| SNPs | Type of Mutation | Genotypes | Number of Samples | BW/cm | FL/cm | BL/cm | BH/cm |
|---|---|---|---|---|---|---|---|
| scaffold12-12716321 | T > C | CC | 101 | 12.89 ± 8.25 b | 8.98 ± 1.88 b | 7.46 ± 1.58 b | 2.82 ± 0.70 |
| CT | 51 | 14.15 ± 7.93 ab | 9.33 ± 1.87 ab | 7.74 ± 1.62 ab | 2.91 ± 0.67 | ||
| TT | 6 | 19.58 ± 6.26 a | 10.45 ± 1.32 a | 8.80 ± 1.12 a | 3.27 ± 0.53 | ||
| scaffold13-4787950 | A > C | AA | 29 | 16.13 ± 7.35 a | 9.72 ± 1.74 a | 8.10 ± 1.46 a | 3.04 ± 0.60 |
| AC | 65 | 13.78 ± 9.14 ab | 9.18 ± 1.99 ab | 7.62 ± 1.69 ab | 2.88 ± 0.74 | ||
| CC | 64 | 12.14 ± 7.19 b | 8.85 ± 1.76 b | 7.36 ± 1.51 b | 2.77 ± 0.64 | ||
| scaffold168-178120 | A > G | AA | 28 | 15.67 ± 7.60 | 9.71 ± 1.79 a | 8.02 ± 1.49 a | 3.01 ± 0.62 |
| AG | 76 | 13.74 ± 7.39 | 9.24 ± 1.76 ab | 7.68 ± 1.51 ab | 2.89 ± 0.66 | ||
| GG | 54 | 12.19 ± 9.27 | 8.73 ± 2.00 b | 7.28 ± 1.71 b | 2.75 ± 0.74 | ||
| scaffold184-115262 | A > G | AA | 1 | 4.17 ± 0.00 b | 6.40 ± 0.00 b | 5.20 ± 0.00 b | 1.90 ± 0.00 b |
| AG | 14 | 13.35 ± 7.88 a | 9.09 ± 1.98 a | 7.51 ± 1.63 a | 2.86 ± 0.72 a | ||
| GG | 143 | 13.63 ± 8.19 a | 9.17 ± 1.86 a | 7.63 ± 1.58 a | 2.87 ± 0.68 a | ||
| scaffold2-13687576 | A > T | AA | 2 | 4.87 ± 1.18 b | 6.7 ± 0.81 b | 5.75 ± 0.52 b | 2.15 ± 0.17 b |
| AT | 34 | 10.96 ± 6.82 ab | 8.63 ± 1.71 a | 7.09 ± 1.41 a | 2.67 ± 0.61 ab | ||
| TT | 122 | 14.41 ± 8.35 a | 9.33 ± 1.88 a | 7.77 ± 1.60 a | 2.93 ± 0.69 a | ||
| scaffold290-11890 | T > C | CC | 19 | 9.46 ± 4.34 b | 8.30 ± 1.29 b | 6.87 ± 1.07 b | 2.53 ± 0.47 b |
| CT | 76 | 14.26 ± 8.77 a | 9.31 ± 1.97 a | 7.74 ± 1.69 a | 2.92 ± 0.72 a | ||
| TT | 63 | 13.92 ± 7.98 a | 9.20 ± 1.86 a | 7.65 ± 1.56 a | 2.90 ± 0.67 a | ||
| scaffold59-960994 | A > G | AA | 1 | 4.09 ± 0.00 b | 6.50 ± 0.00 b | 5.50 ± 0.00 b | 1.08 ± 0.00 b |
| AG | 33 | 15.21 ± 10.30 a | 9.48 ± 2.13 a | 7.89 ± 1.81 a | 2.97 ± 0.75 a | ||
| GG | 124 | 13.18 ± 7.46 a | 9.08 ± 1.79 a | 7.54 ± 1.52 a | 2.85 ± 0.65 a | ||
| scaffold66-172323 | A > G | AA | 3 | 8.44 ± 6.04 | 8.03 ± 1.93 | 6.67 ± 1.46 | 2.19 ± 1.16 b |
| AG | 41 | 14.89 ± 6.85 | 9.53 ± 1.61 | 7.90 ± 1.37 | 3.05 ± 0.58 a | ||
| GG | 114 | 13.20 ± 8.58 | 9.04 ± 1.95 | 7.52 ± 1.66 | 2.81 ± 0.69 ab |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Lin, Z.; Wei, M.; Chen, Z.; Shen, M.; Cao, G.; Wang, Y.; Zhang, Z.; Zhang, D. Development of Single Nucleotide Polymorphism and Association Analysis with Growth Traits for Black Porgy (Acanthopagrus schlegelii). Genes 2022, 13, 1992. https://doi.org/10.3390/genes13111992
Zhang Z, Lin Z, Wei M, Chen Z, Shen M, Cao G, Wang Y, Zhang Z, Zhang D. Development of Single Nucleotide Polymorphism and Association Analysis with Growth Traits for Black Porgy (Acanthopagrus schlegelii). Genes. 2022; 13(11):1992. https://doi.org/10.3390/genes13111992
Chicago/Turabian StyleZhang, Zhiwei, Zhijie Lin, Mingliang Wei, Ziqiang Chen, Mingjun Shen, Guangyong Cao, Yue Wang, Zhiyong Zhang, and Dianchang Zhang. 2022. "Development of Single Nucleotide Polymorphism and Association Analysis with Growth Traits for Black Porgy (Acanthopagrus schlegelii)" Genes 13, no. 11: 1992. https://doi.org/10.3390/genes13111992
APA StyleZhang, Z., Lin, Z., Wei, M., Chen, Z., Shen, M., Cao, G., Wang, Y., Zhang, Z., & Zhang, D. (2022). Development of Single Nucleotide Polymorphism and Association Analysis with Growth Traits for Black Porgy (Acanthopagrus schlegelii). Genes, 13(11), 1992. https://doi.org/10.3390/genes13111992