
A Novel De Novo NFKBIA Missense Mutation Associated to Ectodermal Dysplasia with Dysgammaglobulinemia
 , , , , and
, , , , and
Abstract
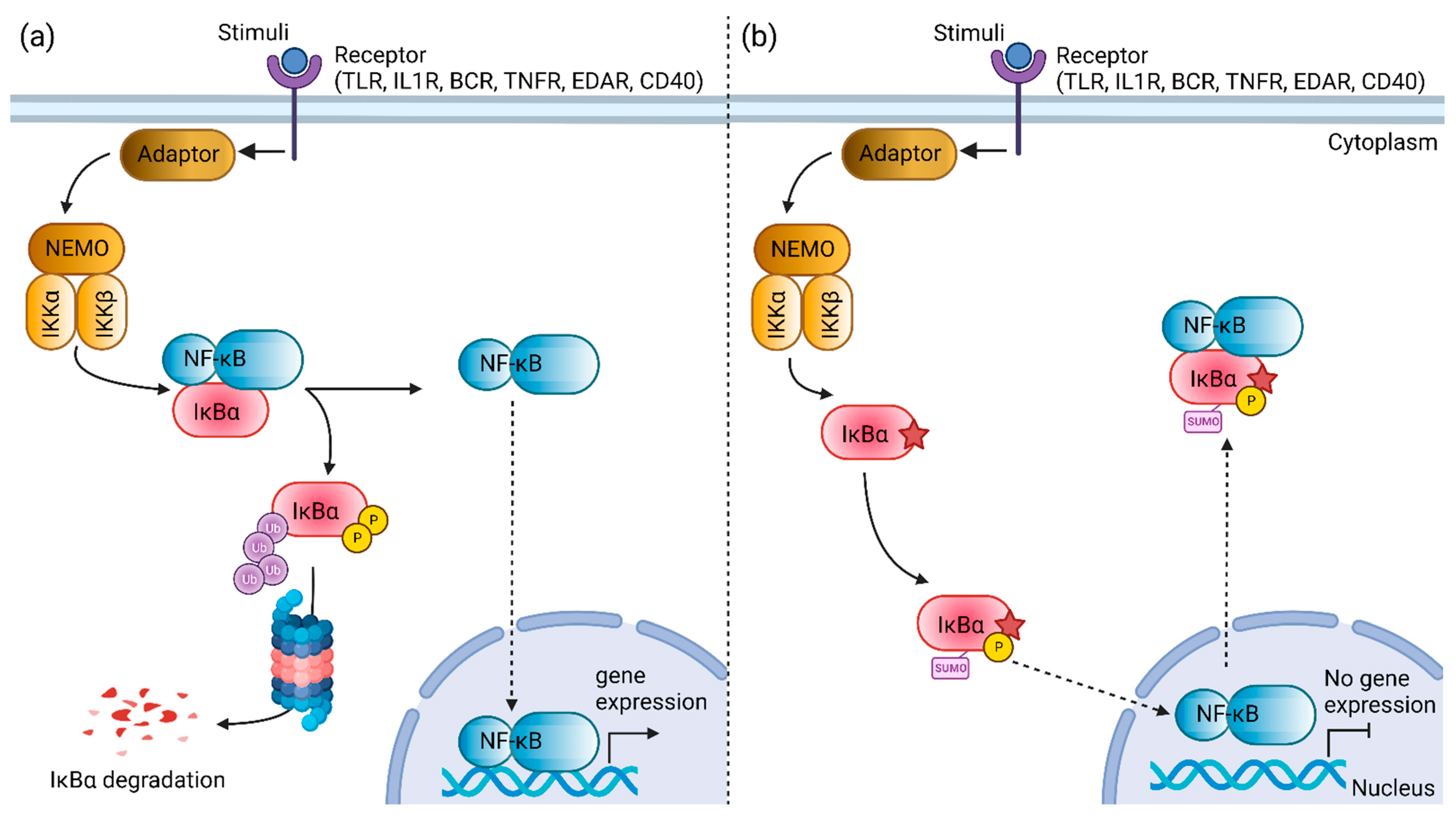
1. Introduction
2. Materials and Methods
2.1. Patient
2.2. Whole-Exome Sequencing and Bioinformatics Analysis
2.3. Variant Validation via Sanger Sequencing
2.4. Conservation Analysis
2.5. Structural Effect Evaluation of Ser32 Variants
2.6. Phosphorylation Prediction
2.7. Post-Translational Modification Site Prediction
3. Results
3.1. Clinical Features
3.2. Bioinformatics Analysis
3.3. Structural Effect Evaluation of Ser32 Variants
3.4. Post-Translational Modification Site Predictions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bousfiha, A.; Jeddane, L.; Picard, C.; Al-Herz, W.; Ailal, F.; Chatila, T.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; Holland, S.M.; et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J. Clin. Immunol. 2020, 40, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Cunningham-Rundles, C.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; Oksenhendler, E.; Picard, C.; et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2022, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Engelbrecht, C.; Urban, M.; Schoeman, M.; Paarwater, B.; van Coller, A.; Abraham, D.R.; Cornelissen, H.; Glashoff, R.; Esser, M.; Möller, M.; et al. Clinical Utility of Whole Exome Sequencing and Targeted Panels for the Identification of Inborn Errors of Immunity in a Resource-Constrained Setting. Front. Immunol. 2021, 12, 665621. [Google Scholar] [CrossRef]
- Meyts, I.; Bosch, B.; Bolze, A.; Boisson, B.; Itan, Y.; Belkadi, A.; Pedergnana, V.; Moens, L.; Picard, C.; Cobat, A.; et al. Exome and Genome Sequencing for Inborn Errors of Immunity. J. Allergy Clin. Immunol. 2016, 138, 957–969. [Google Scholar] [CrossRef]
- Ripen, A.M.; Chear, C.T.; Baharin, M.F.; Nallusamy, R.; Chan, K.C.; Kassim, A.; Choo, C.M.; Wong, K.J.; Fong, S.M.; Tan, K.K.; et al. A Single-center Pilot Study in Malaysia on the Clinical Utility of Whole-exome Sequencing for Inborn Errors of Immunity. Clin. Exp. Immunol. 2021, 206, 119–128. [Google Scholar] [CrossRef] [PubMed]
- García-Martín, P.; Hernández-Martín, A.; Torrelo, A. Ectodermal Dysplasias: A Clinical and Molecular Review. Actas Dermo-Sifiliográficas 2013, 104, 451–470. [Google Scholar] [CrossRef]
- Cardinez, C.; Miraghazadeh, B.; Tanita, K.; da Silva, E.; Hoshino, A.; Okada, S.; Chand, R.; Asano, T.; Tsumura, M.; Yoshida, K.; et al. Gain-of-Function IKBKB Mutation Causes Human Combined Immune Deficiency. J. Exp. Med. 2018, 215, 2715–2724. [Google Scholar] [CrossRef]
- Kawai, T.; Nishikomori, R.; Heike, T. Diagnosis and Treatment in Anhidrotic Ectodermal Dysplasia with Immunodeficiency. Allergol. Int. 2012, 61, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Zinatizadeh, M.R.; Schock, B.; Chalbatani, G.M.; Zarandi, P.K.; Jalali, S.A.; Miri, S.R. The Nuclear Factor Kappa B (NF-KB) Signaling in Cancer Development and Immune Diseases. Genes Dis. 2021, 8, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Etzioni, A.; Ochs, H.D. The Hyper IgM Syndrome—An Evolving Story. Pediatr. Res. 2004, 56, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Boisson, B.; Puel, A.; Picard, C.; Casanova, J.-L. Human IκBα Gain of Function: A Severe and Syndromic Immunodeficiency. J. Clin. Immunol. 2017, 37, 397–412. [Google Scholar] [CrossRef]
- Batlle-Masó, L. Genetic Diagnosis of Autoinflammatory Disease Patients Using Clinical Exome Sequencing. Eur. J. Med. Genet. 2020, 63, 103920. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Wang, L.; Deng, M.; Li, Y.; Tang, X.; Mao, H.; Zhao, X. A Heterozygous N-Terminal Truncation Mutation of NFKBIA Results in an Impaired NF-ΚB Dependent Inflammatory Response. Genes Dis. 2022, 9, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Chang, X.; Wang, K. WANNOVAR: Annotating Genetic Variants for Personal Genomes via the Web. J. Med. Genet. 2012, 49, 433–436. [Google Scholar] [CrossRef]
- Chear, C.T.; Nallusamy, R.; Canna, S.W.; Chan, K.C.; Baharin, M.F.; Hishamshah, M.; Ghani, H.; Ripen, A.M.; Mohamad, S.B. A Novel de Novo NLRC4 Mutation Reinforces the Likely Pathogenicity of Specific LRR Domain Mutation. Clin. Immunol. 2020, 211, 108328. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the Deleteriousness of Variants throughout the Human Genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Söding, J.; Thompson, J.D.; et al. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Venselaar, H.; te Beek, T.A.; Kuipers, R.K.; Hekkelman, M.L.; Vriend, G. Protein Structure Analysis of Mutations Causing Inheritable Diseases. An e-Science Approach with Life Scientist Friendly Interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef]
- Wagih, O.; Reimand, J.; Bader, G.D. MIMP: Predicting the Impact of Mutations on Kinase-Substrate Phosphorylation. Nat. Methods 2015, 12, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zeng, S.; Xu, C.; Qiu, W.; Liang, Y.; Joshi, T.; Xu, D. MusiteDeep: A Deep-Learning Framework for General and Kinase-Specific Phosphorylation Site Prediction. Bioinformatics 2017, 33, 3909–3916. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liu, D.; Yuchi, J.; He, F.; Jiang, Y.; Cai, S.; Li, J.; Xu, D. MusiteDeep: A Deep-Learning Based Webserver for Protein Post-Translational Modification Site Prediction and Visualization. Nucleic Acids Res. 2020, 48, W140–W146. [Google Scholar] [CrossRef] [PubMed]
- Prashanth, S.; Deshmukh, S. Ectodermal Dysplasia: A Genetic Review. Int. J. Clin. Pediatr. Dent. 2012, 5, 197–202. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-ΚB Pathway for the Therapy of Diseases: Mechanism and Clinical Study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef]
- Truhlar, S.M.E.; Torpey, J.W.; Komives, E.A. Regions of IkBa That Are Critical for Its Inhibition of NF-KB.DNA Interaction Fold upon Binding to NF-KB. Proc. Natl. Acad. Sci. USA 2006, 103, 18951–18956. [Google Scholar] [CrossRef]
- Wang, X.; Peng, H.; Huang, Y.; Kong, W.; Cui, Q.; Du, J.; Jin, H. Post-Translational Modifications of IκBα: The State of the Art. Front. Cell Dev. Biol. 2020, 8, 574706. [Google Scholar] [CrossRef]
- Yazdi, S.; Naumann, M.; Stein, M. Double Phosphorylation-Induced Structural Changes in the Signal-Receiving Domain of IκBα in Complex with NF-ΚB: Double-Phosphorylation Events in the SRD of IκBα. Proteins 2017, 85, 17–29. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Kanarek, N.; Ben-Neriah, Y. Regulation of NF-ΚB by Ubiquitination and Degradation of the IκBs: IκB Ubiquitination and Degradation. Immunol. Rev. 2012, 246, 77–94. [Google Scholar] [CrossRef]
- Courtois, G.; Smahi, A.; Reichenbach, J.; Döffinger, R.; Cancrini, C.; Bonnet, M.; Puel, A.; Chable-Bessia, C.; Yamaoka, S.; Feinberg, J.; et al. A Hypermorphic IκBα Mutation Is Associated with Autosomal Dominant Anhidrotic Ectodermal Dysplasia and T Cell Immunodeficiency. J. Clin. Investig. 2003, 112, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Sogkas, G.; Adriawan, I.R.; Ringshausen, F.C.; Baumann, U.; Schröder, C.; Klemann, C.; von Hardenberg, S.; Schmidt, G.; Bernd, A.; Jablonka, A.; et al. A Novel NFKBIA Variant Substituting Serine 36 of IκBα Causes Immunodeficiency with Warts, Bronchiectasis and Juvenile Rheumatoid Arthritis in the Absence of Ectodermal Dysplasia. Clin. Immunol. 2020, 210, 108269. [Google Scholar] [CrossRef] [PubMed]
- Dupuis-Girod, S.; Cancrini, C.; Le Deist, F.; Palma, P.; Bodemer, C.; Puel, A.; Livadiotti, S.; Picard, C.; Bossuyt, X.; Rossi, P.; et al. Successful Allogeneic Hemopoietic Stem Cell Transplantation in a Child Who Had Anhidrotic Ectodermal Dysplasia With Immunodeficiency. Pediatrics 2006, 118, e205–e211. [Google Scholar] [CrossRef] [PubMed]
- Giancane, G.; Ferrari, S.; Carsetti, R.; Papoff, P.; Iacobini, M.; Duse, M. Anhidrotic Ectodermal Dysplasia: A New Mutation. J. Allergy Clin. Immunol. 2013, 132, 1451–1453. [Google Scholar] [CrossRef]
- Janssen, R.; van Wengen, A.; Hoeve, M.A.; ten Dam, M.; van der Burg, M.; van Dongen, J.; van de Vosse, E.; van Tol, M.; Bredius, R.; Ottenhoff, T.H.; et al. The Same IκBα Mutation in Two Related Individuals Leads to Completely Different Clinical Syndromes. J. Exp. Med. 2004, 200, 559–568. [Google Scholar] [CrossRef]
- Schimke, L.F.; Rieber, N.; Rylaarsdam, S.; Cabral-Marques, O.; Hubbard, N.; Puel, A.; Kallmann, L.; Sombke, S.A.; Notheis, G.; Schwarz, H.-P.; et al. A Novel Gain-of-Function IKBA Mutation Underlies Ectodermal Dysplasia with Immunodeficiency and Polyendocrinopathy. J. Clin. Immunol. 2013, 33, 1088–1099. [Google Scholar] [CrossRef]
- Staples, E.; Morillo-Gutierrez, B.; Davies, J.; Petersheim, D.; Massaad, M.; Slatter, M.; Dimou, D.; Doffinger, R.; Hackett, S.; Kumararatne, D.; et al. Disseminated Mycobacterium Malmoense and Salmonella Infections Associated with a Novel Variant in NFKBIA. J. Clin. Immunol. 2017, 37, 415–418. [Google Scholar] [CrossRef]
- Moriya, K.; Sasahara, Y.; Ohnishi, H.; Kawai, T.; Kanegane, H. IKBA S32 Mutations Underlie Ectodermal Dysplasia with Immunodeficiency and Severe Noninfectious Systemic Inflammation. J. Clin. Immunol. 2018, 38, 543–545. [Google Scholar] [CrossRef]
- Petersheim, D.; Massaad, M.J.; Lee, S.; Scarselli, A.; Cancrini, C.; Moriya, K.; Sasahara, Y.; Lankester, A.C.; Dorsey, M.; Di Giovanni, D.; et al. Mechanisms of Genotype-Phenotype Correlation in Autosomal Dominant Anhidrotic Ectodermal Dysplasia with Immune Deficiency. J. Allergy Clin. Immunol. 2018, 141, 1060–1073.e3. [Google Scholar] [CrossRef]
- Chen, Z.; Hagler, J.; Palombella, V.J.; Melandri, F.; Scherer, D.; Ballard, D.; Maniatis, T. Signal-Induced Site-Specific Phosphorylation Targets I Kappa B Alpha to the Ubiquitin-Proteasome Pathway. Genes Dev. 1995, 9, 1586–1597. [Google Scholar] [CrossRef]
- Yaron, A.; Hatzubai, A.; Davis, M.; Lavon, I.; Amit, S.; Manning, A.M.; Andersen, J.S.; Mann, M.; Mercurio, F.; Ben-Neriah, Y. Identifcation of the Receptor Component of the IkBa Ubiquitin Ligase. Nature 1998, 396, 5. [Google Scholar] [CrossRef] [PubMed]
- Celen, A.B.; Sahin, U. Sumoylation on Its 25th Anniversary: Mechanisms, Pathology, and Emerging Concepts. FEBS J. 2020, 287, 3110–3140. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Post-Translational Modifications Regulating the Activity and Function of the Nuclear Factor Kappa B Pathway. Oncogene 2006, 25, 6717–6730. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-Y.; Li, F.C.H.; Wu, C.H.Y.; Chang, A.Y.W.; Chan, S.H.H. Sumoylation of IkB Attenuates NF-KB-Induced Nitrosative Stress at Rostral Ventrolateral Medulla and Cardiovascular Depression in Experimental Brain Death. J. Biomed. Sci. 2016, 23, 65. [Google Scholar] [CrossRef]
- Cánovas, D.; Marcos, J.F.; Marcos, A.T.; Strauss, J. Nitric Oxide in Fungi: Is There NO Light at the End of the Tunnel? Curr. Genet 2016, 62, 513–518. [Google Scholar] [CrossRef]
- Henard, C.A.; Vázquez-Torres, A. Nitric Oxide and Salmonella Pathogenesis. Front. Microbio. 2011, 2, 84. [Google Scholar] [CrossRef]
- Wiegand, S.B.; Traeger, L.; Nguyen, H.K.; Rouillard, K.R.; Fischbach, A.; Zadek, F.; Ichinose, F.; Schoenfisch, M.H.; Carroll, R.W.; Bloch, D.B.; et al. Antimicrobial Effects of Nitric Oxide in Murine Models of Klebsiella Pneumonia. Redox Biol. 2021, 39, 101826. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immunological Parameters | Result | Reference Range |
|---|---|---|
| Lymphocyte subsets (Absolute count, ×106 cells/L) | ||
| Total T cells | 11,529 | 1700–3600 |
| Total B cells | 1099 | 500–1500 |
| CD4 cells | 9555 | 1700–2800 |
| CD8 cells | 3647 | 800–1200 |
| NK cells | 223 | 300–700 |
| Immunoglobulins | ||
| IgG (g/L) | 0.91 | 2.6–15.2 |
| IgA (g/L) | 0.08 | 0.16–1.1 |
| IgM (g/L) | 5.85 | 0.10–1.2 |
| IgE (kU/L) | 4.43 | <7.3 |
| Complements (g/L) | ||
| C3 | 1.55 | 0.5–0.9 |
| C4 | 0.29 | 0.1–0.4 |
| Gene | Mutation | Phosphorylation Site | Name of Kinase | Family of Kinase | Joint Probability * | Effect |
|---|---|---|---|---|---|---|
| NFKBIA | Ser32Cys | 32 | CHUK | IKK | 0.9997 | loss |
| NFKBIA | Ser32Cys | 32 | TGFBR2 | STKR_Type2 | 0.9829 | loss |
| NFKBIA | Ser32Cys | 32 | IKBKE | IKK | 0.9809 | loss |
| NFKBIA | Ser32Cys | 32 | PRKACA | PKA | 0.9375 | loss |
| NFKBIA | Ser32Cys | 32 | IKBKB | IKK | 0.9338 | loss |
| NFKBIA | Ser32Cys | 32 | PRKAA1 | CAMKL_AMPK | 0.8779 | loss |
| NFKBIA | Ser32Cys | 32 | CSNK2A1 | CK2 | 0.8514 | loss |
| NFKBIA | Ser32Cys | 32 | DAPK3 | DAPK_DAPK | 0.8496 | loss |
| NFKBIA | Ser32Cys | 32 | PRKCE | PKC_Eta | 0.8392 | loss |
| NFKBIA | Ser32Cys | 32 | CAMK1 | CAMK1 | 0.8206 | loss |
| NFKBIA | Ser32Cys | 32 | PRKD1 | PKD | 0.8176 | loss |
| NFKBIA | Ser32Cys | 32 | RPS6KB1 | RSK_p70 | 0.8101 | loss |
| NFKBIA | Ser32Cys | 32 | MAP3K8 | STE-Unique | 0.8001 | loss |
| NFKBIA | Ser32Cys | 32 | CAMK2A | CAMK2 | 0.7852 | loss |
| NFKBIA | Ser32Cys | 32 | CHEK1 | CAMKL_CHK1 | 0.7832 | loss |
| NFKBIA | Ser32Cys | 32 | CAMK4 | CAMK1 | 0.7608 | loss |
| NFKBIA | Ser32Cys | 32 | SGK1 | SGK | 0.7565 | loss |
| NFKBIA | Ser32Cys | 32 | PRKAA2 | CAMKL_AMPK | 0.6896 | loss |
| NFKBIA | Ser32Cys | 32 | AKT2 | Akt | 0.6866 | loss |
| NFKBIA | Ser32Cys | 32 | PAK2 | STE20_PAKA | 0.6696 | loss |
| NFKBIA | Ser32Cys | 32 | RPS6KA3 | RSK_RSK | 0.6637 | loss |
| NFKBIA | Ser32Cys | 32 | PRKCD | PKC_Delta | 0.6406 | loss |
| NFKBIA | Ser32Cys | 32 | PIM1 | PIM | 0.5352 | loss |
| NFKBIA | Ser32Cys | 32 | CSNK2A2 | CK2 | 0.5298 | loss |
| NFKBIA | Ser32Cys | 32 | PAK1 | STE20_PAKA | 0.5292 | loss |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chear, C.T.; El Farran, B.A.K.; Sham, M.; Ramalingam, K.; Noh, L.M.; Ismail, I.H.; Chiow, M.Y.; Baharin, M.F.; Ripen, A.M.; Mohamad, S.B. A Novel De Novo NFKBIA Missense Mutation Associated to Ectodermal Dysplasia with Dysgammaglobulinemia. Genes 2022, 13, 1900. https://doi.org/10.3390/genes13101900
Chear CT, El Farran BAK, Sham M, Ramalingam K, Noh LM, Ismail IH, Chiow MY, Baharin MF, Ripen AM, Mohamad SB. A Novel De Novo NFKBIA Missense Mutation Associated to Ectodermal Dysplasia with Dysgammaglobulinemia. Genes. 2022; 13(10):1900. https://doi.org/10.3390/genes13101900
Chicago/Turabian StyleChear, Chai Teng, Bader Abdul Kader El Farran, Marina Sham, Kavetha Ramalingam, Lokman Mohd Noh, Intan Hakimah Ismail, Mei Yee Chiow, Mohd Farid Baharin, Adiratna Mat Ripen, and Saharuddin Bin Mohamad. 2022. "A Novel De Novo NFKBIA Missense Mutation Associated to Ectodermal Dysplasia with Dysgammaglobulinemia" Genes 13, no. 10: 1900. https://doi.org/10.3390/genes13101900
APA StyleChear, C. T., El Farran, B. A. K., Sham, M., Ramalingam, K., Noh, L. M., Ismail, I. H., Chiow, M. Y., Baharin, M. F., Ripen, A. M., & Mohamad, S. B. (2022). A Novel De Novo NFKBIA Missense Mutation Associated to Ectodermal Dysplasia with Dysgammaglobulinemia. Genes, 13(10), 1900. https://doi.org/10.3390/genes13101900