
Optimization of Oxford Nanopore Technology Sequencing Workflow for Detection of Amplicons in Real Time Using ONT-DART Tool
 , ,
, ,
Abstract
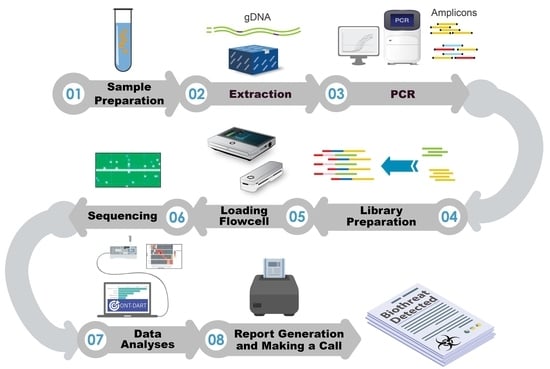
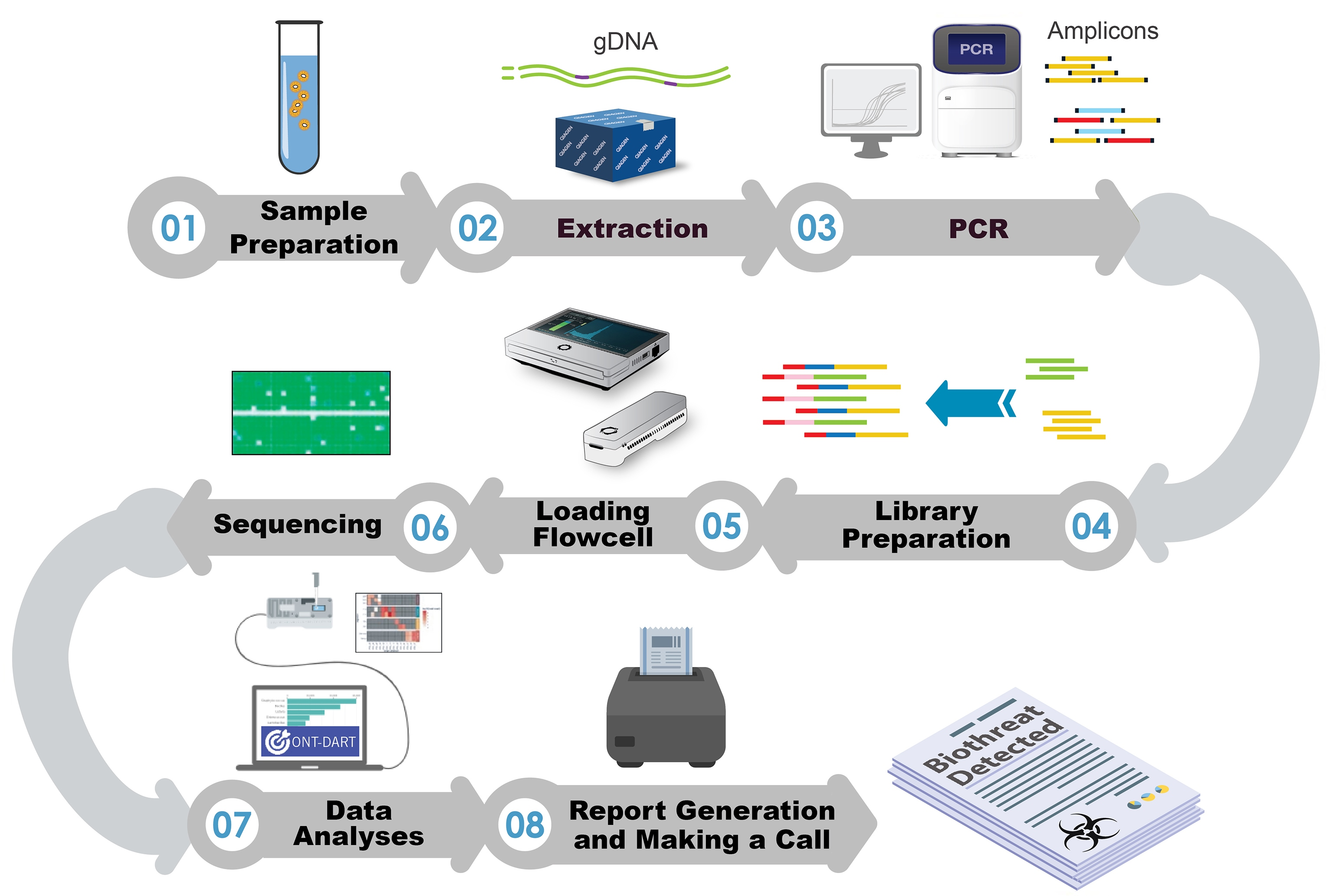
1. Introduction
2. Materials and Methods
2.1. Sample Preparation and PCR Amplification
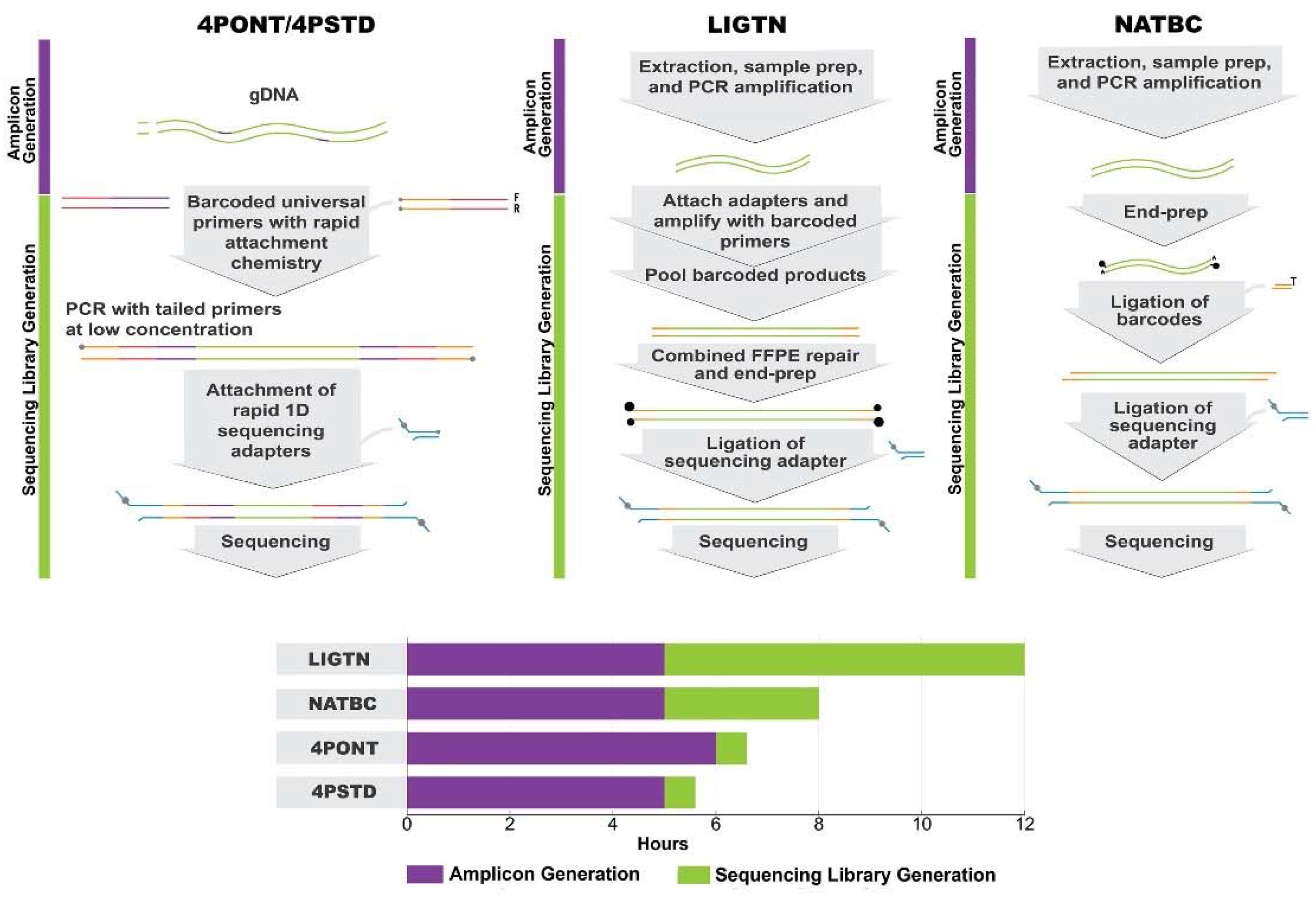
2.2. Library Preparation Method 1: 4PSTD
2.3. Library Preparation Method 2: 4PONT–4-Primer Modified
2.4. Library Method 3: NATBC–Native Barcoding
2.5. Library Method 4: LIGTN-Ligation
2.6. ONT Sequencing
2.7. Post-Sequence Processing and Analysis
3. Results and Discussion
3.1. Description of Amplification Strategies
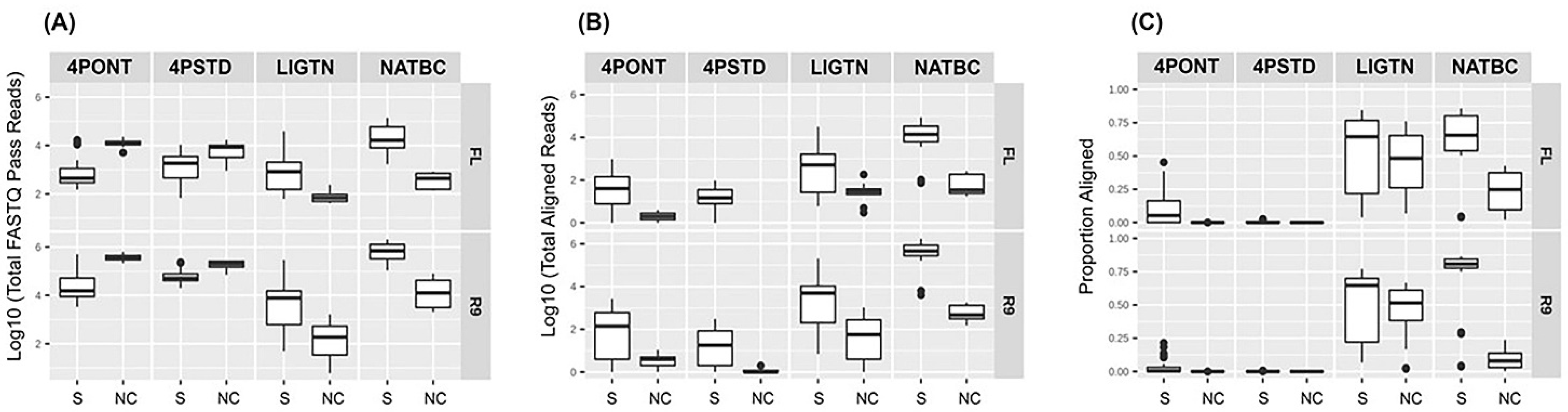
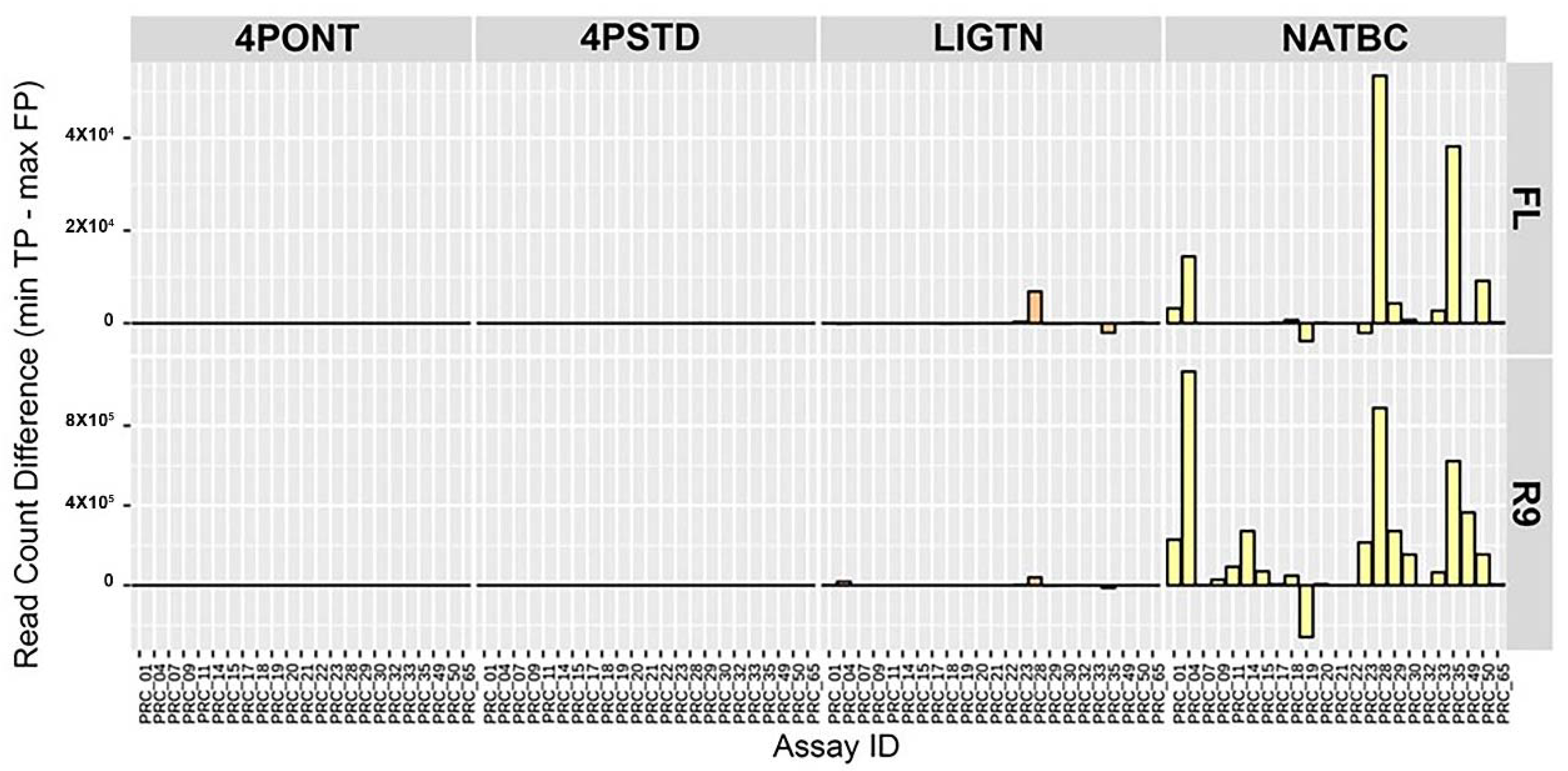
3.2. Comparison of Library Preparation Strategies
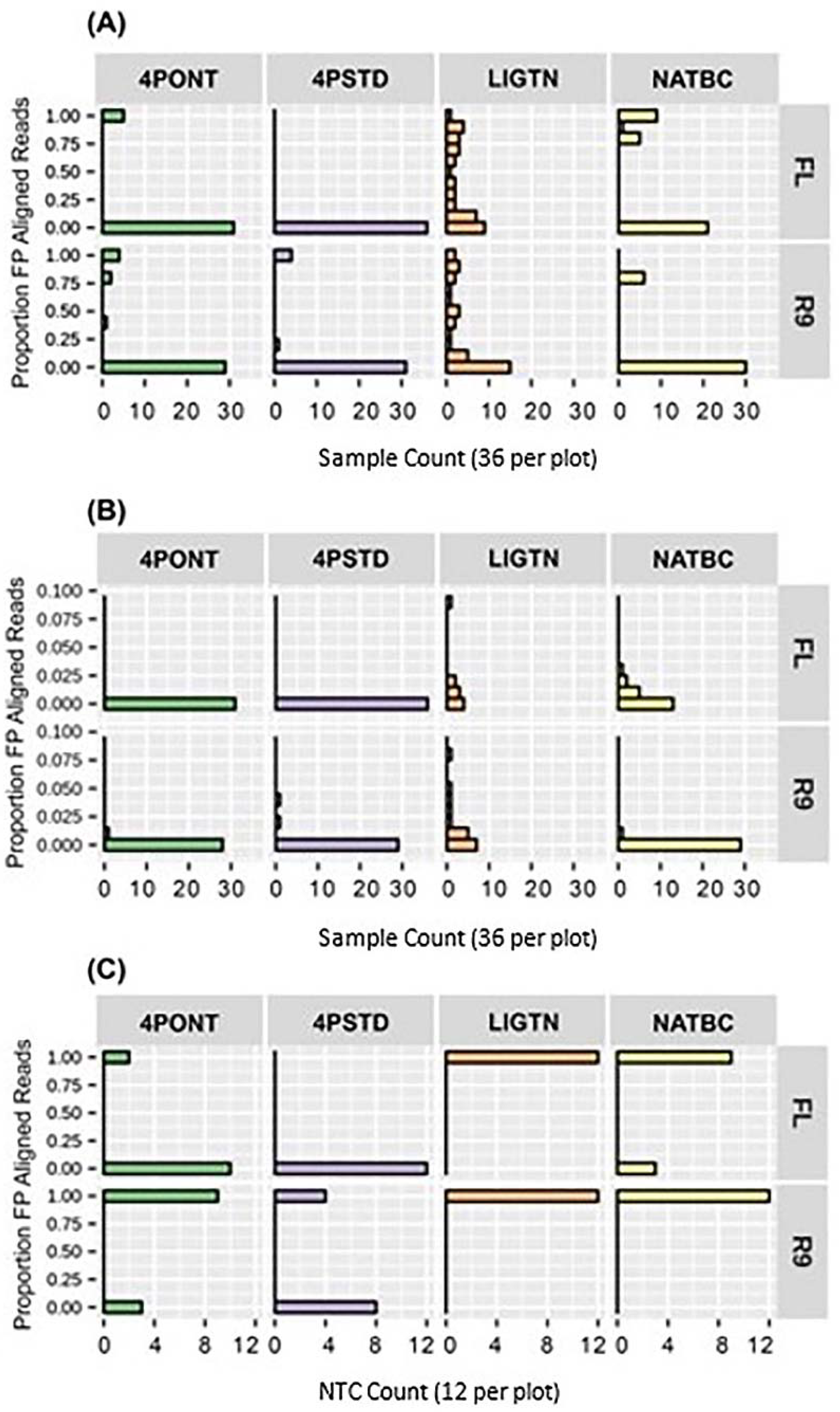
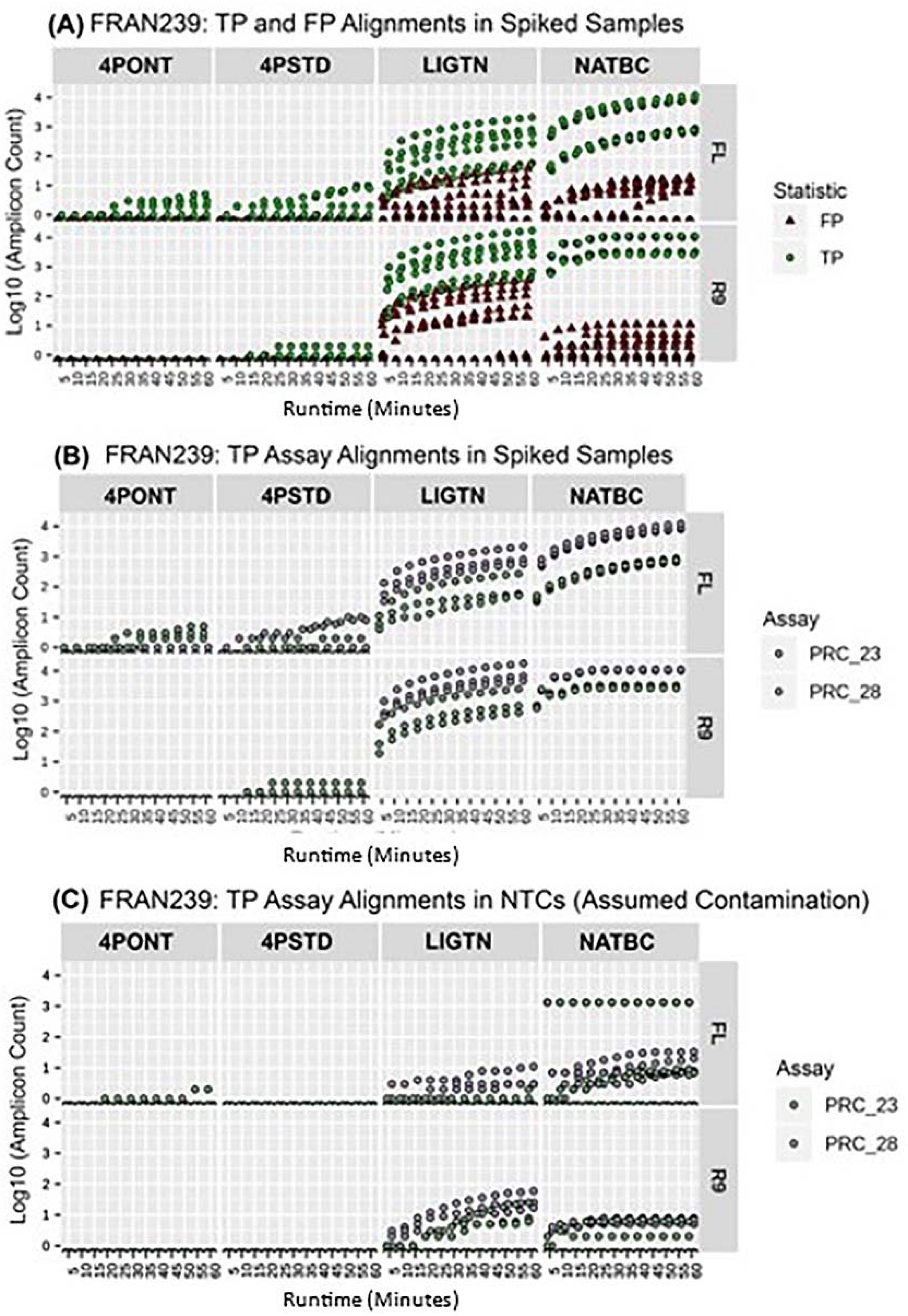
3.3. Analysis of NTCs
3.4. ONT-DART Analysis Pipeline
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef] [PubMed]
- Zeller, M.; Gangavarapu, K.; Anderson, C.; Smither, A.R.; Vanchiere, J.A.; Rose, R.; Snyder, D.J.; Dudas, G.; Watts, A.; Matteson, N.L.; et al. Emergence of an early SARS-CoV-2 epidemic in the United States. Cell 2021, 184, 4939–4952.e15. [Google Scholar] [CrossRef] [PubMed]
- Minogue, T.D.; Koehler, J.W.; Stefan, C.P.; Conrad, T.A. Next-Generation Sequencing for Biodefense: Biothreat Detection, Forensics, and the Clinic. Clin. Chem. 2019, 65, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Conrad, T.A.; Lo, C.C.; Koehler, J.W.; Graham, A.S.; Stefan, C.P.; Hall, A.T.; Douglas, C.E.; Chain, P.S.; Minogue, T.D. Diagnostic targETEd seQuencing adjudicaTion (DETEQT): Algorithms for Adjudicating Targeted Infectious Disease Next-Generation Sequencing Panels. J. Mol. Diagn. 2019, 21, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, S.V.; Reed, T.M.; Sullivan, R.F.; Kerkhof, L.J.; Beigel, K.M.; Wade, M.M. Offline Next Generation Metagenomics Sequence Analysis Using MinION Detection Software (MINDS). Genes 2019, 10, 578. [Google Scholar] [CrossRef] [PubMed]
- Colman, R.E.; Schupp, J.M.; Hicks, N.D.; Smith, D.E.; Buchhagen, J.L.; Valafar, F.; Crudu, V.; Romancenco, E.; Noroc, E.; Jackson, L.; et al. Detection of Low-Level Mixed-Population Drug Resistance in Mycobacterium tuberculosis Using High Fidelity Amplicon Sequencing. PLoS ONE 2015, 10, e0126626. [Google Scholar] [CrossRef] [PubMed]
- Calus, S.T.; Ijaz, U.Z.; Pinto, A.J. NanoAmpli-Seq: A workflow for amplicon sequencing for mixed microbial communities on the nanopore sequencing platform. Gigascience 2018, 7, giy140. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chen, Q.; Xiong, M.; Zhao, J.; Shen, S.; Chen, L.; Pan, Y.; Li, Z.; Li, Y. Clinical Performance of Nanopore Targeted Sequencing for Diagnosing Infectious Diseases. Microbiol. Spectr. 2022, 10, e0027022. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.C.; Wu, H.C.; Liou, C.H.; Lauderdale, T.L.; Huang, I.W.; Lai, J.F.; Chen, F.J. Rapid and Routine Molecular Typing Using Multiplex Polymerase Chain Reaction and MinION Sequencer. Front. Microbiol. 2022, 13, 875347. [Google Scholar] [CrossRef] [PubMed]
- Player, R.; Verratti, K.; Staab, A.; Bradburne, C.; Grady, S.; Goodwin, B.; Sozhamannan, S. Comparison of the performance of an amplicon sequencing assay based on Oxford Nanopore technology to real-time PCR assays for detecting bacterial biodefense pathogens. BMC Genomics 2020, 21, 166. [Google Scholar] [CrossRef] [PubMed]
- Myers, W.G.; Altschul, S.F.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- GNU Parallel: The Command-Line Power Tool|USENIX. Available online: https://www.usenix.org/publications/login/february-2011-volume-36-number-1/gnu-parallel-command-line-power-tool (accessed on 1 September 2021).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar] [CrossRef]
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 1 September 2021).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method Name | 4-Primer ONT | 4-Primer Standard | Ligation | Native Barcoding | |
|---|---|---|---|---|---|
| Method ID | 4PONT | 4PSTD | LIGTN | NATBC | |
| PCR Step | Standard Targeted PCR | x | x | ||
| 4P PCR (ONT cycling parameters) | x | ||||
| 4P PCR (custom cycling parameters) | x | ||||
| Library Prep Step | End prep and cleanup | x | x | ||
| Ligate barcode adapter and cleanup | x | ||||
| Attach barcode with PCR | x | ||||
| NB ligation and cleanup | x | ||||
| Pool barcoded libraries | x | x | |||
| End prep and ligate seq adapter | x | x | x | x | |
| Load pooled library on to flow cell | x | x | x | x | |
| Time (hours) | Sample preparation | 2 | 2 | 2 | 2 |
| gDNA extraction | 2 | 2 | 2 | 2 | |
| PCR amplification | 0 | 0 | 1 | 1 | |
| Library prep | 2 | 1 | 7 | 3 | |
| Total time until sequencing | 6 | 5 | 12 | 8 | |
| No. | Organism ID | Organism | Strain | Expected PRC Assays |
|---|---|---|---|---|
| 1 | BANT708 | Bacillus anthracis | Sterne BAP708 | 01, 04, 07 |
| 2 | BCER248 | Bacillus cereus | NRS 248 | 07 |
| 3 | BRUC105 | Brucella abortus | RB51 | 32, 33, 35 |
| 4 | BRUC106 | Brucella abortus | Strain 19 | 32, 33, 35 |
| 5 | BURK164 | Burkholderia humptydooensis | MSMB121 | 49 |
| 6 | BURK197 | Burkholderia pseudomallei | JW270 | 50, 65 |
| 7 | FRAN239 | Francisella tularensis | NIH B-38 | 23, 28 |
| 8 | FRAN240 | Francisella tularensis | LVS | 23, 29 |
| 9 | FRAN241 | Francisella tularensis | Novidica U112 | 23, 30 |
| 10 | VACCIN | Vaccinia | 17, 18, 20 | |
| 11 | YERS113 | Yersinia pestis | CO92 Lcr (-) | 09, 11, 15 |
| 12 | YERS114 | Yersinia pestis | CO92 pgm (-) | 09, 14, 15 |
| Minutes until All TP Amplicons > 9 Reads | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Device | Flongle Flow Cell | R9 Flow Cell | |||||||
| Method | 4PONT | 4PSTD | LIGTN | NATBC | 4PONT | 4PSTD | LIGTN | NATBC | |
| Organism ID | BANT708 | >60 | >60 | >60 | 10 | >60 | >60 | 15 | 5 |
| BCER248 | >60 | >60 | >60 | >60 | >60 | >60 | >60 | >60 | |
| BRUC105 | >60 | >60 | >60 | >60 | >60 | >60 | >60 | >60 | |
| BRUC106 | >60 | >60 | >60 | >60 | >60 | >60 | >60 | >60 | |
| BURK164 | >60 | >60 | >60 | >60 | >60 | >60 | >60 | 5 | |
| BURK197 | >60 | >60 | >60 | 45 | >60 | >60 | >60 | 10 | |
| FRAN239 | >60 | >60 | 25 | 5 | >60 | >60 | 5 | 5 | |
| FRAN240 | >60 | >60 | 35 | 5 | >60 | >60 | 10 | 5 | |
| FRAN241 | >60 | >60 | 50 | 5 | >60 | >60 | 10 | 5 | |
| VACCIN | >60 | >60 | >60 | >60 | >60 | >60 | >60 | 5 | |
| YERS113 | >60 | >60 | >60 | >60 | >60 | >60 | >60 | 5 | |
| YERS114 | >60 | >60 | >60 | >60 | >60 | >60 | >60 | 10 | |
| Min | >60 | >60 | 25 | 10 | >60 | >60 | 5 | 5 | |
| Max | >60 | >60 | >60 | >60 | >60 | >60 | >60 | >60 | |
| Method | 4PONT | 4PSTD | LIGTN | NATBC | |||||
|---|---|---|---|---|---|---|---|---|---|
| NTC/Sample | NTC | Sample | NTC | Sample | NTC | Sample | NTC | Sample | |
| Mean | 0 (14) | 0 (0) | 0 (0) | 0 (0) | 204 (38) | 41 (44) | 2547 (39) | 70 (22) | FL |
| Median | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 26 (25) | 30 (48) | 247 (2) | 24 (25) | |
| Standard Deviation | 1 (35) | 1 (0) | 0 (0) | 0 (0) | 598 (35) | 47 (24) | 4162 (46) | 96 (16) | |
| Mean | 1 (17) | 3 (0) | 0 (12) | 0 (0) | 1266 (30) | 202 (44) | 19447 (13) | 753 (9) | R9 |
| Median | 0 (0) | 3 (0) | 0 (0) | 0 (0) | 94 (11) | 84 (52) | 350 (0) | 464 (8) | |
| Standard Deviation | 4 (36) | 4 (0) | 1 (32) | 1 (0) | 3343 (36) | 309 (24) | 63551 (29) | 622 (8) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Player, R.; Verratti, K.; Staab, A.; Forsyth, E.; Ernlund, A.; Joshi, M.S.; Dunning, R.; Rozak, D.; Grady, S.; Goodwin, B.; et al. Optimization of Oxford Nanopore Technology Sequencing Workflow for Detection of Amplicons in Real Time Using ONT-DART Tool. Genes 2022, 13, 1785. https://doi.org/10.3390/genes13101785
Player R, Verratti K, Staab A, Forsyth E, Ernlund A, Joshi MS, Dunning R, Rozak D, Grady S, Goodwin B, et al. Optimization of Oxford Nanopore Technology Sequencing Workflow for Detection of Amplicons in Real Time Using ONT-DART Tool. Genes. 2022; 13(10):1785. https://doi.org/10.3390/genes13101785
Chicago/Turabian StylePlayer, Robert, Kathleen Verratti, Andrea Staab, Ellen Forsyth, Amanda Ernlund, Mihir S. Joshi, Rebecca Dunning, David Rozak, Sarah Grady, Bruce Goodwin, and et al. 2022. "Optimization of Oxford Nanopore Technology Sequencing Workflow for Detection of Amplicons in Real Time Using ONT-DART Tool" Genes 13, no. 10: 1785. https://doi.org/10.3390/genes13101785
APA StylePlayer, R., Verratti, K., Staab, A., Forsyth, E., Ernlund, A., Joshi, M. S., Dunning, R., Rozak, D., Grady, S., Goodwin, B., & Sozhamannan, S. (2022). Optimization of Oxford Nanopore Technology Sequencing Workflow for Detection of Amplicons in Real Time Using ONT-DART Tool. Genes, 13(10), 1785. https://doi.org/10.3390/genes13101785