
Insights into the Host-Pathogen Interaction Pathways through RNA-Seq Analysis of Lens culinaris Medik. in Response to Rhizoctonia bataticola Infection

 ,
,  , , , , , and
, , , , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Fungal Inoculation
2.2. RNA Extraction, cDNA Library Construction, and Illumina RNA-Sequencing
2.3. De Novo Transcriptome Assembly
2.4. Transcriptome Annotation and Gene Ontology (GO)
2.5. Differential Gene Expression Analysis
2.6. GO-ENRICHMENT and Pathway Analysis
2.7. PHI-Base Analysis
2.8. Identification of miRNA and miRNA Target Sites
2.9. Protein-Protein Interaction (PPI) Network
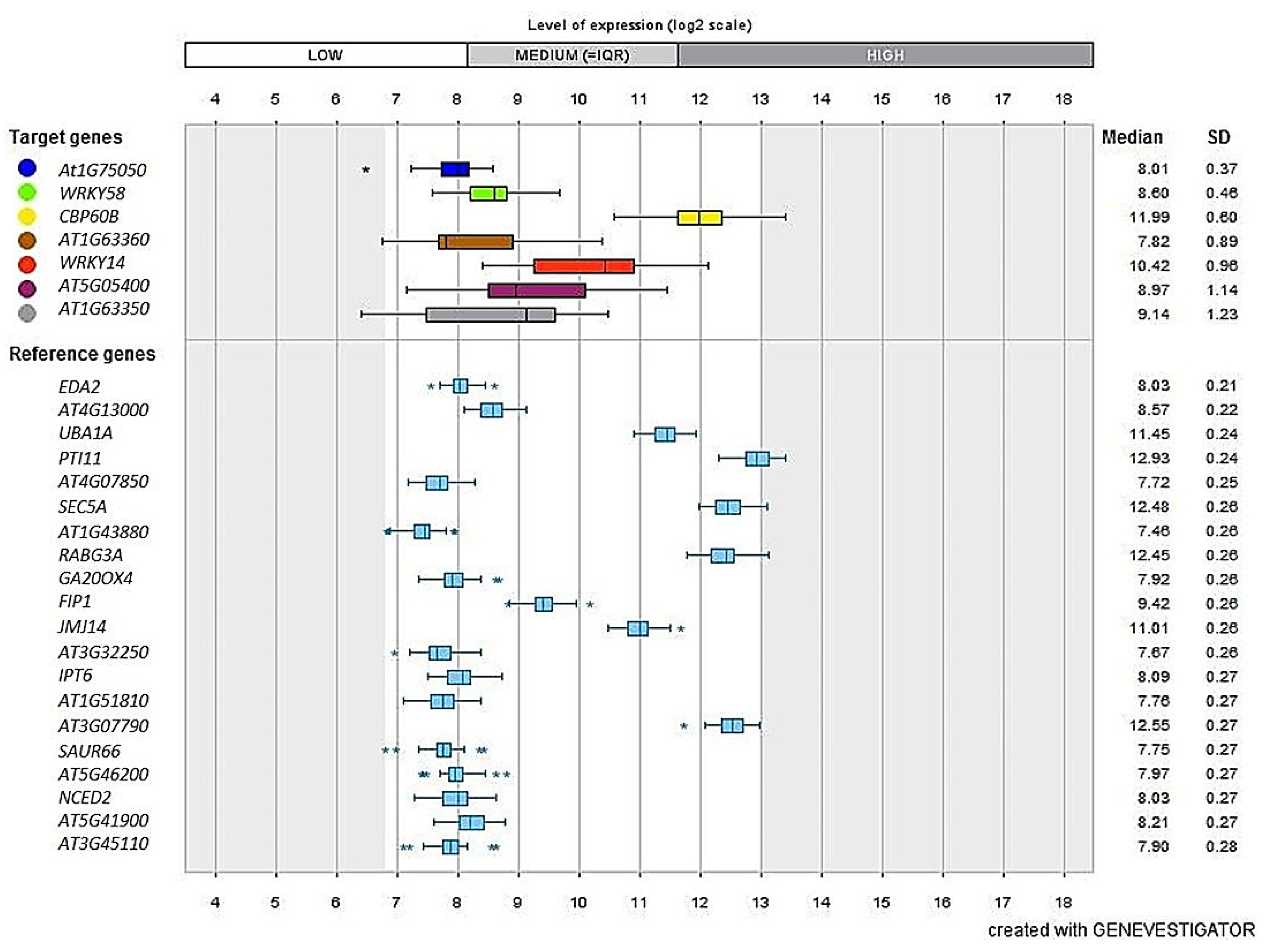
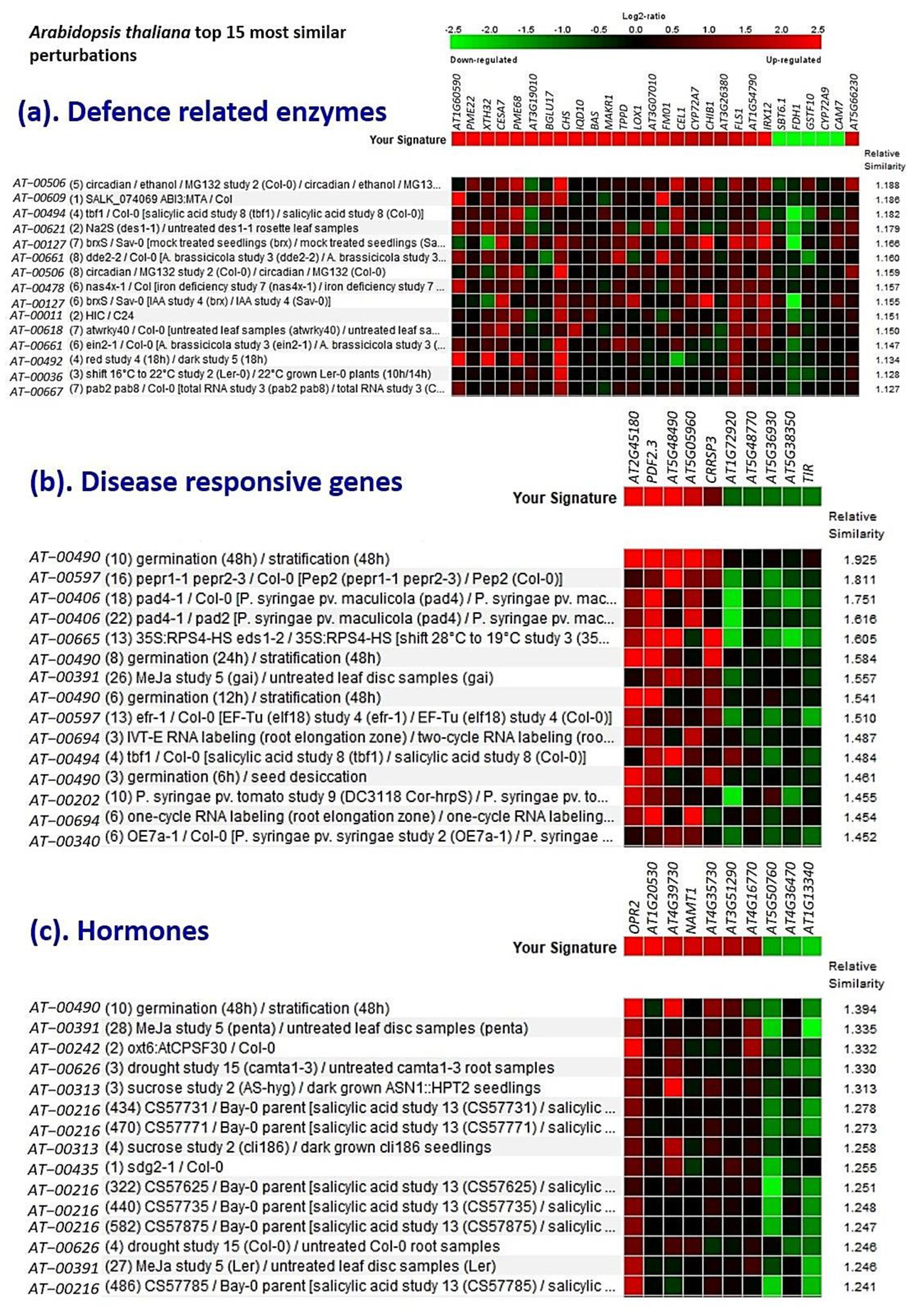
2.10. InSilico Validation Analysis Using Genevestigator®
3. Results
3.1. Fungus Confirmation and Inoculation
3.2. RNA Sequencing and De Novo Transcriptome Assembly
3.3. Differential Gene Expression Analysis
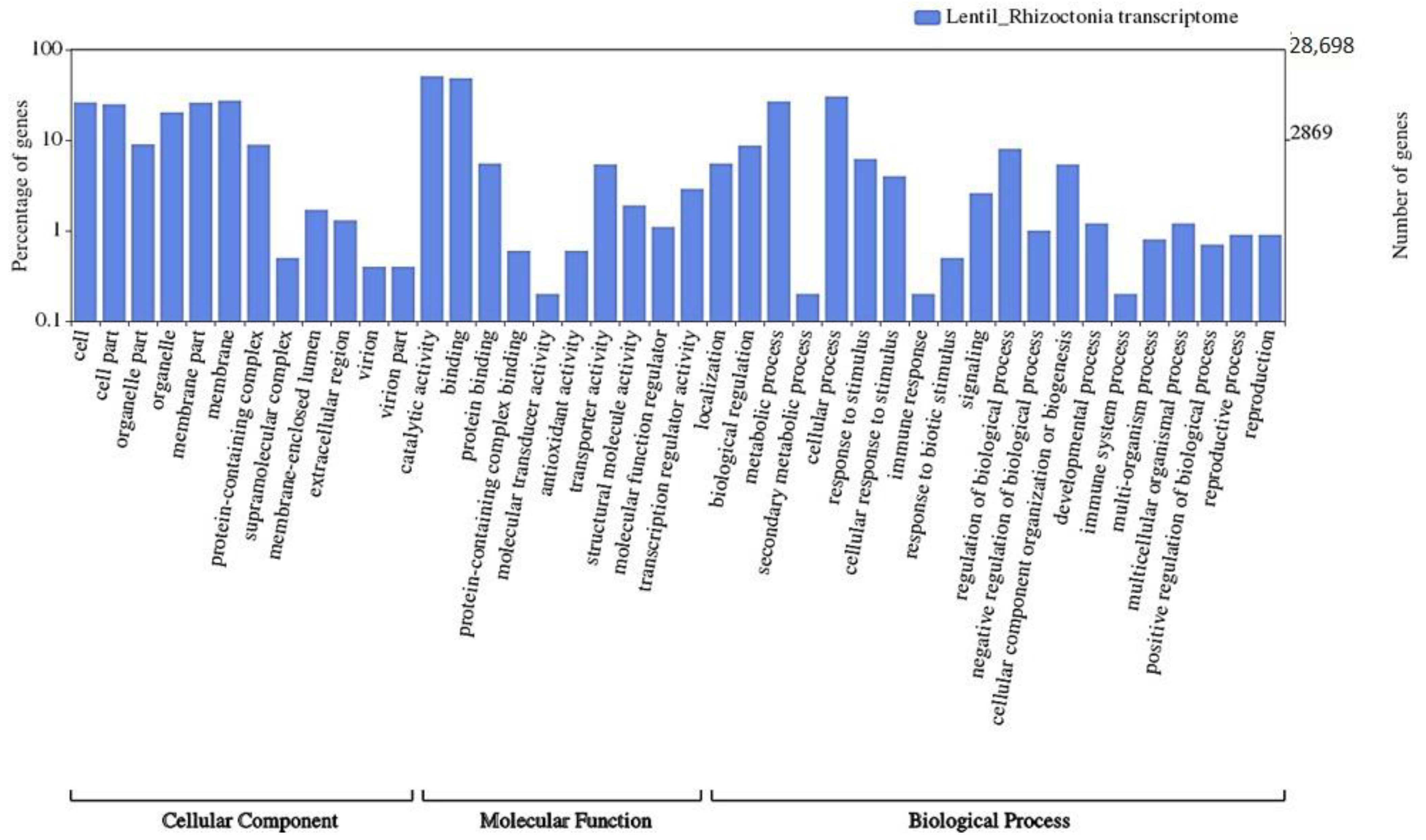
3.4. Transcriptome Annotation
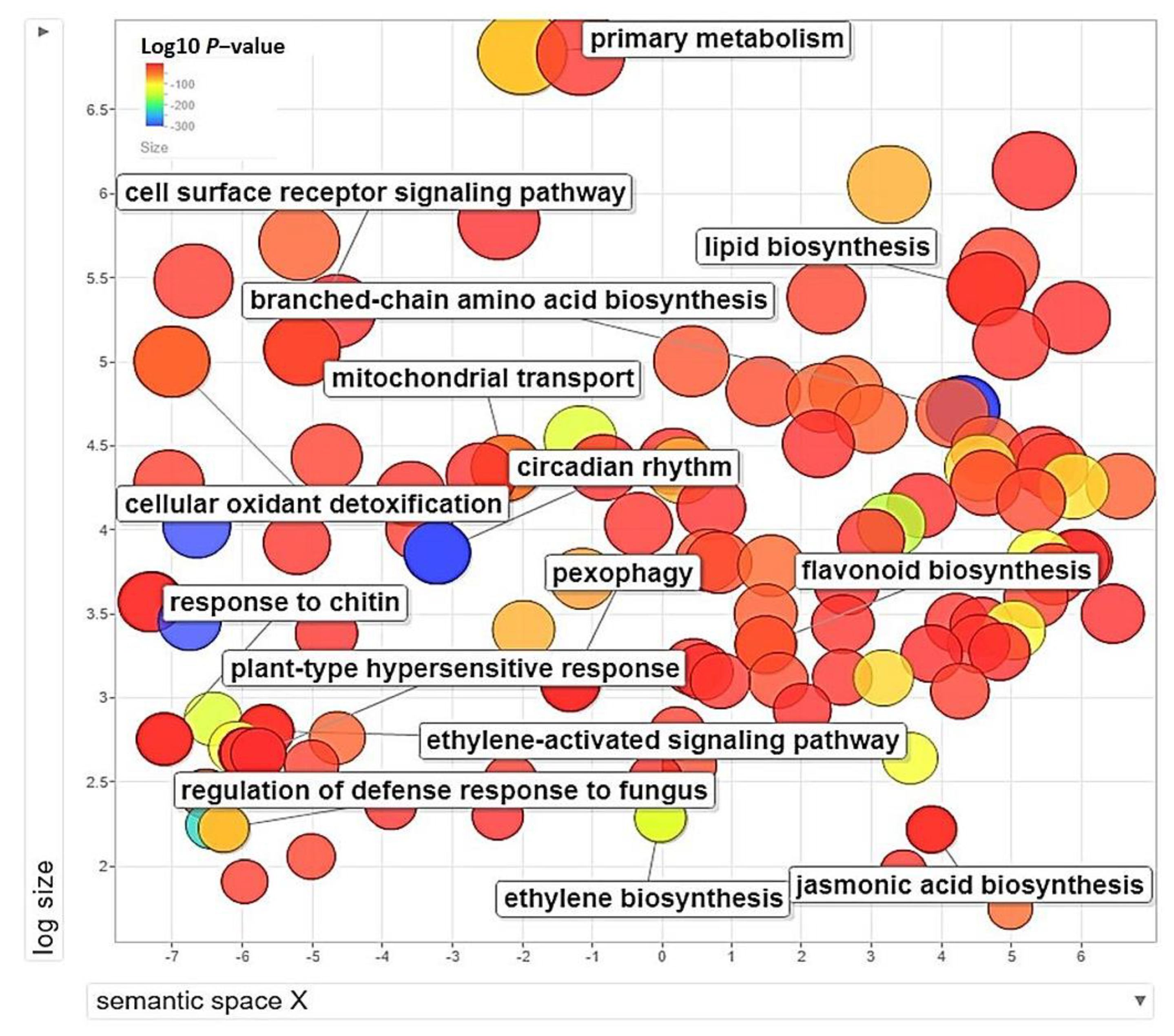
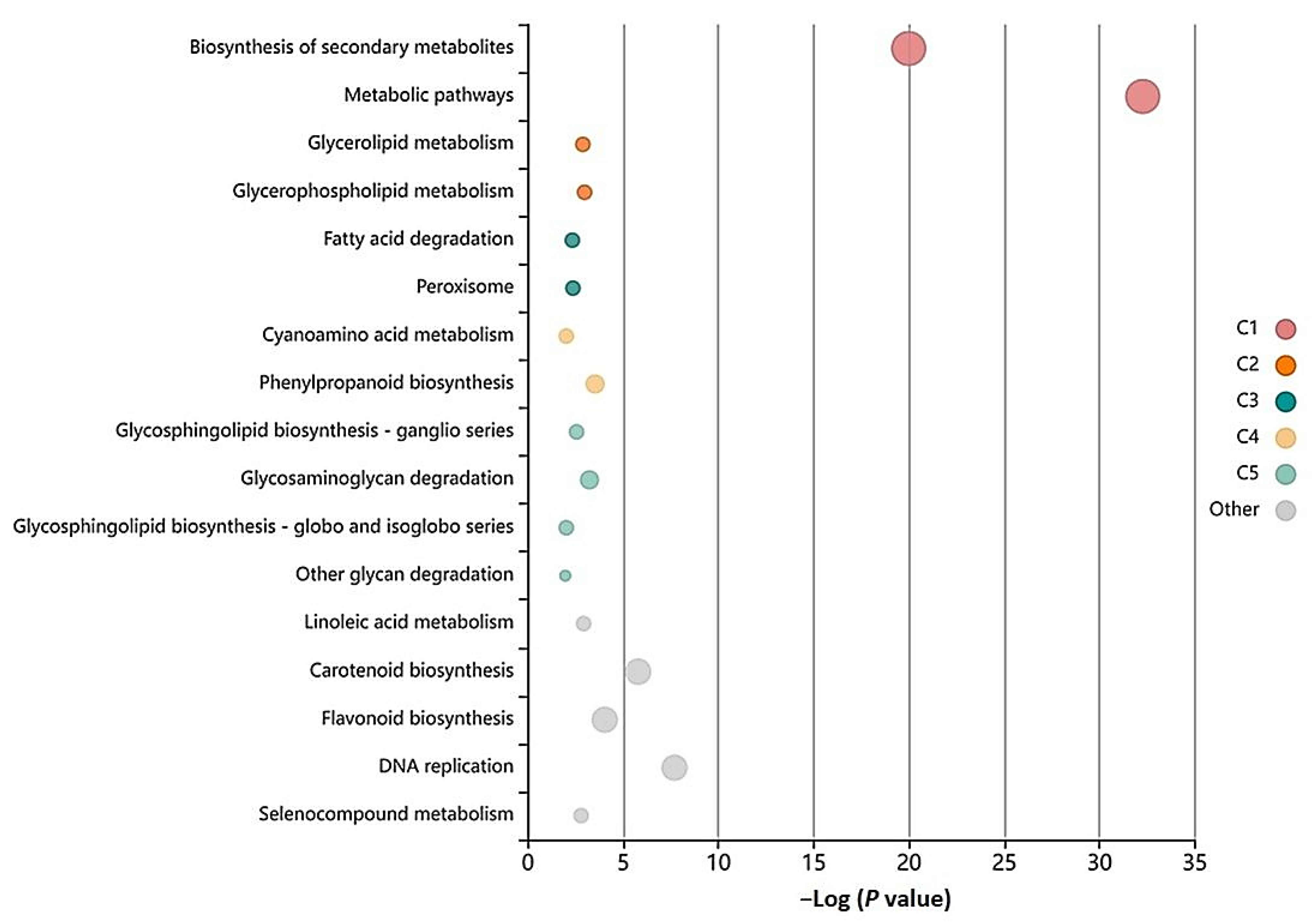
3.5. GO-Enrichment and Pathway Analysis
3.6. PHI-Base: Pathogen Host Interaction
3.7. miRNA Prediction, miRNA Target Site Identification, and Functional Analysis
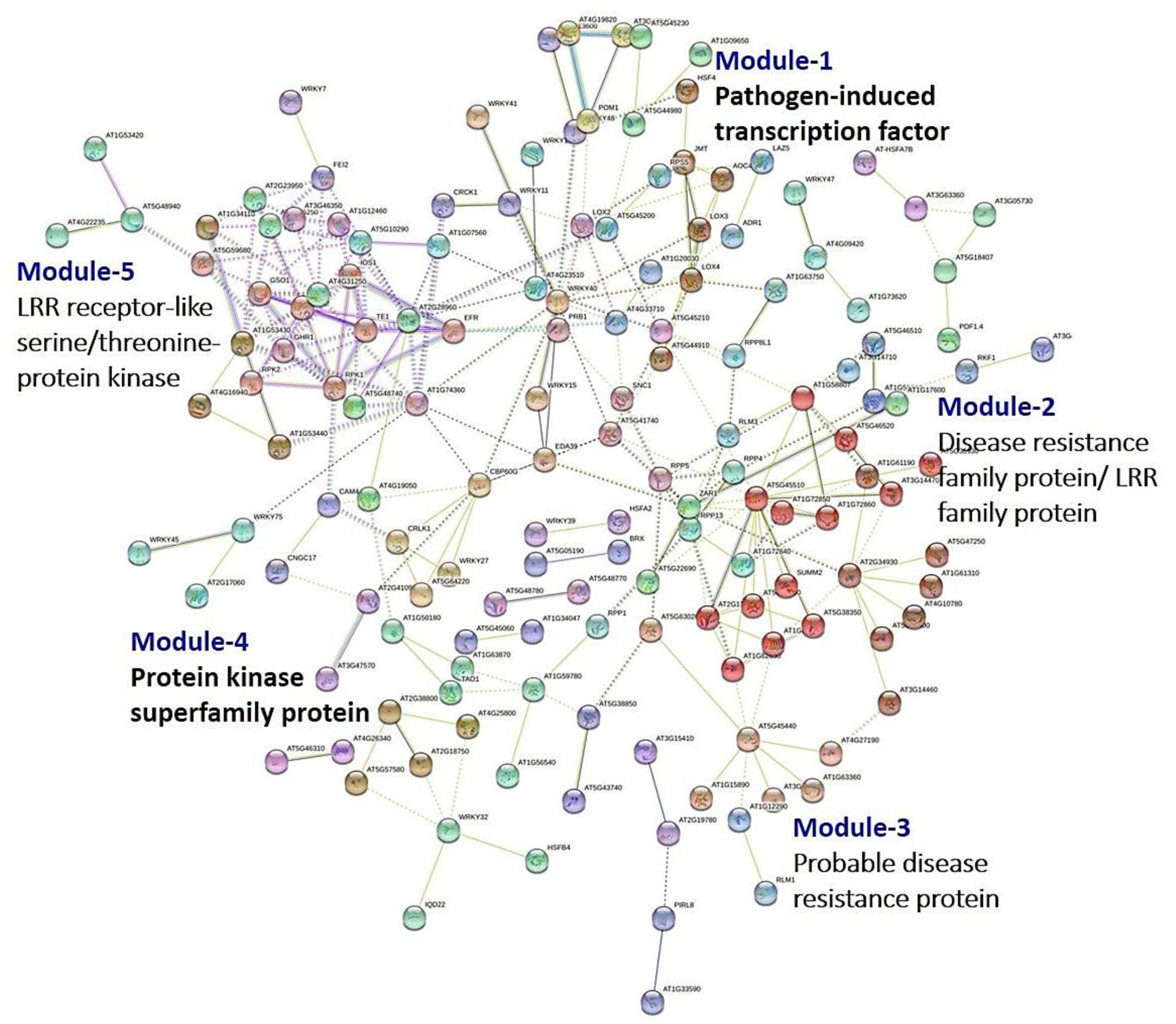
3.8. PPI Network Analysis
3.9. Validation Studies Using InSilico Expression Analysis
4. Discussion
4.1. Gene Expression and Validation
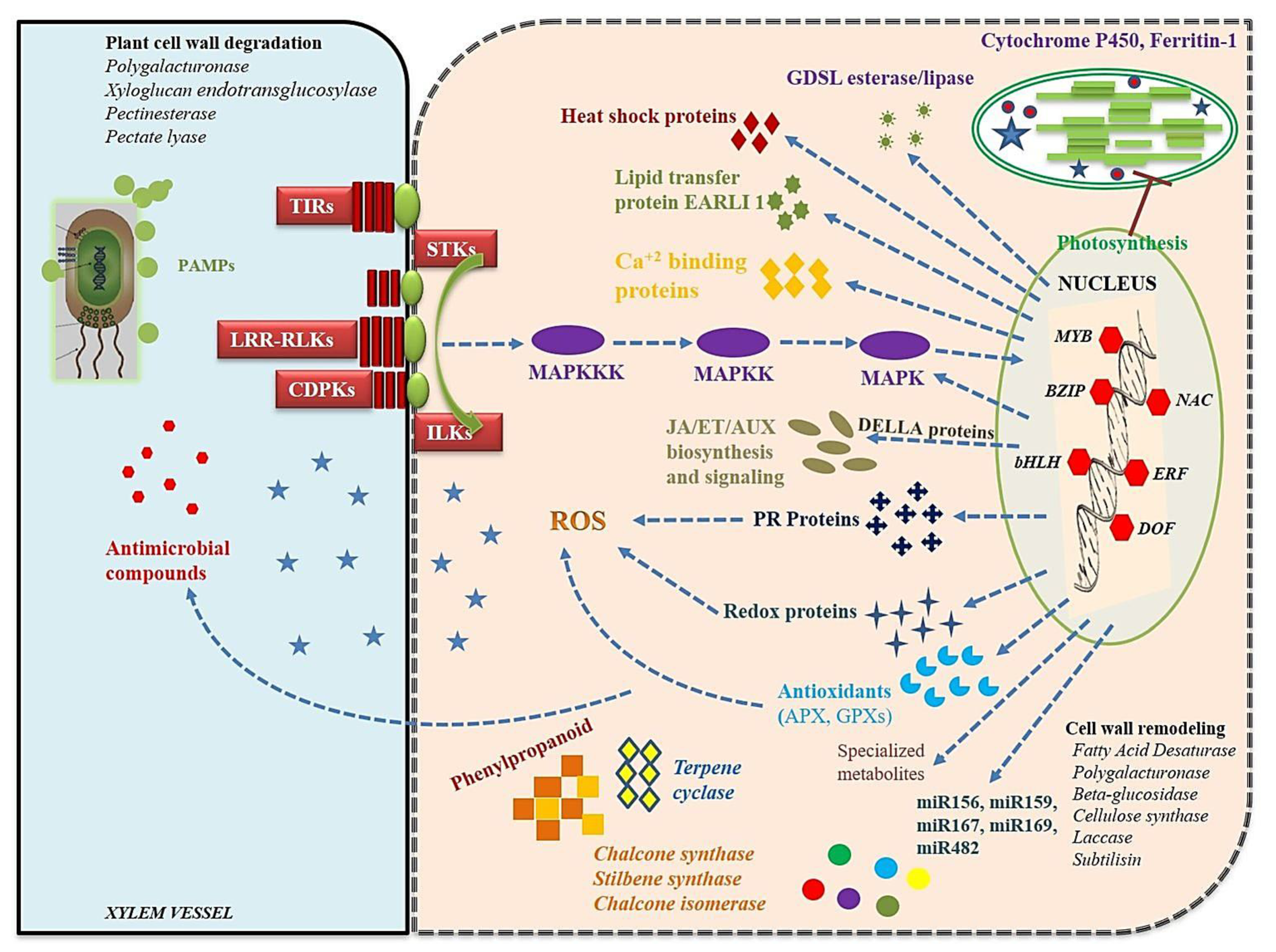
4.2. Recognition of Rhizoctonia and Expression of Plant Signaling Pathways
4.3. Structural and Biochemical Response to Rhizoctonia Infection
4.4. Hypersensitive Response (HR), Cell Death, and Systemic Acquired Resistance (SAR)
4.5. TFs Related to Fungal RESPONSES
4.6. Role of miRNAs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arumuganathan, K.; Earle, E.D. Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep. 1991, 9, 208–218. [Google Scholar] [CrossRef]
- Kumar, S.; Rajendran, K.; Kumar, J.; Hamwieh, A.; Baum, M. Current knowledge in lentil genomics and its application for crop improvement. Front. Plant Sci. 2015, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- Roy, F.; Boye, J.; Simpson, B. Bioactive proteins and peptides in pulse crops: Pea, chickpea and lentil. Food Res. Int. 2010, 43, 432–442. [Google Scholar] [CrossRef]
- Mishra, G.P.; Dikshit, H.K.; Tontang, M.T.; Stobdan, T.; Sangwan, S.; Aski, M.; Singh, A.; Kumar, R.R.; Tripathi, K.; Kumar, S.; et al. Diversity in phytochemical composition, antioxidant capacities, and nutrient contents among mungbean and lentil microgreens when grown at plain-altitude region (Delhi) and high-altitude region (Leh-Ladakh), India. Front. Plant Sci. 2021, 12, 710812. [Google Scholar] [CrossRef]
- Mishra, G.P.; Dikshit, H.K.; Kumari, J.; Priti; Tripathi, K.; Devi, J.; Aski, M.; Mehra, R.; Sarker, A.; Kumar, S. Identification and characterization of novel penta-podded genotypes in the cultivated lentil (Lens culinaris Medik.). Crop. Sci. 2020, 60, 1974–1985. [Google Scholar] [CrossRef]
- Mishra, G.P.; Dikshit, H.K.; Sv, R.; Tripathi, K.; Kumar, R.R.; Aski, M.; Singh, A.; Roy, A.; Kumari, N.; Dasgupta, U.; et al. Yellow Mosaic Disease (YMD) of Mungbean (Vigna radiata (L.) Wilczek): Current Status and Management Opportunities. Front. Plant Sci. 2020, 11, 918. [Google Scholar] [CrossRef]
- Constabel, C.P.; Yoshida, K.; Walker, V. Diverse Ecological Roles of Plant Tannins: Plant Defense and Beyond. Recent Adv. Polyphen. Res. 2014, 4, 115–142. [Google Scholar] [CrossRef]
- Chino, X.M.S.; Martinez, C.J.; Dávila-Ortiz, G.; Alvarez-Gonzalez, I.; Madrigal-Bujaidar, E. Nutrient and Non-nutrient Components of Legumes, and Its Chemopreventive Activity: A Review. Nutr. Cancer 2015, 67, 401–410. [Google Scholar] [CrossRef]
- FAO; FAOSTAT. Food and Agriculture Organization of the United Nations. Rome. 2021. Available online: http://faostat.fao.org (accessed on 9 November 2021).
- Kaur, S.; Dhillon, G.S.; Brar, S.K.; Vallad, G.E.; Chand, R.; Chauhan, V.B. Emerging phytopathogen Macrophomina phaseolina: Biology, economic importance and current diagnostic trends. Crit. Rev. Microbiol. 2011, 38, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.F.; Gossen, B.D.; Chang, K.F.; Turnbull, G.D.; Howard, R.J.; Blade, S.F. Etiology, impact and control of rhizoctonia seedling blight and root rot of chickpea on the Canadian prairies. Can. J. Plant Sci. 2003, 83, 959–967. [Google Scholar] [CrossRef]
- Mengistu, A.; Smith, J.R.; Ray, J.D.; Bellaloui, N. Seasonal progress of charcoal rot and its impact on soybean productivity. Plant Dis. 2011, 95, 1159–1166. [Google Scholar] [CrossRef]
- Živanov, D.; Živanov, S.T.; Nagl, N.; Savić, A.; Katanski, S.; Milić, D. First Report of Macrophomina phaseolina Causing Dry Root Rot of Chickpea (Cicer arietinum) in Serbia. Plant Dis. 2019, 103, 2685. [Google Scholar] [CrossRef]
- Su, G.; Suh, S.-O.; Schneider, R.W.; Russin, J.S. Host Specialization in the Charcoal Rot Fungus, Macrophomina phaseolina. Phytopathology 2001, 91, 120–126. [Google Scholar] [CrossRef]
- Raguchander, T.; Samiyappan, R.; Arjunan, G. Biocontrol of Macrophomina root rot of mungbean. Indian Phytopathol. 1993, 46, 379–382. [Google Scholar]
- Dinakaran, D.; Mohammed, N. Identification of resistant sources to root rot of sesame caused by Macrophomina phaseolina (Tassi.) Goid. Sesame Safflower Newsl. 2001, 16, 68–71. [Google Scholar]
- Kraft, J.M.; Haware, M.P.; Jiménez-Díaz, R.M.; Bayaa, B.; Harrabi, M. Screening techniques and sources of resistance to root rots and wilts in cool season food legumes. Euphytica 1993, 73, 27–39. [Google Scholar] [CrossRef]
- Savary, S.; Nelson, A.; Sparks, A.H.; Willocquet, L.; Duveiller, E.; Mahuku, G.; Forbes, G.; Garrett, K.A.; Hodson, D.; Padgham, J.; et al. International agricultural research tackling the effects of global and climate changes on plant diseases in the develop-ing world. Plant Dis. 2011, 95, 1204–1216. [Google Scholar] [CrossRef]
- Wrather, J.A.; Koenning, S.R.; Anderson, T.R. Effect of Diseases on Soybean Yields in the United States and Ontario (1999 to 2002). Plant Health Prog. 2003, 4, 24. [Google Scholar] [CrossRef]
- Kämper, J.; Kahmann, R.; Bölker, M.; Ma, L.-J.; Brefort, T.; Saville, B.J.; Banuett, F.; Kronstad, J.W.; Gold, S.E.; Müller, O.; et al. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 2006, 444, 97–101. [Google Scholar] [CrossRef]
- Naidoo, S.; Visser, E.A.; Zwart, L.; du Toit, Y.; Bhadauria, V.; Shuey, L. Dual RNA-Sequencing to Elucidate the Plant-Pathogen Duel. Curr. Issues Mol. Biol. 2018, 27, 127–142. [Google Scholar] [CrossRef]
- Phule, A.S.; Barbadikar, K.M.; Maganti, S.M.; Seguttuvel, P.; Subrahmanyam, D.; Babu, M.B.B.P.; Kumar, P.A. RNA-seq reveals the involvement of key genes for aerobic adaptation in rice. Sci. Rep. 2019, 9, 5235. [Google Scholar] [CrossRef]
- Bosamia, T.C.; Dodia, S.M.; Mishra, G.; Ahmad, S.; Joshi, B.; Thirumalaisamy, P.P.; Kumar, N.; Rathnakumar, A.L.; Sangh, C.; Kumar, A.; et al. Unraveling the mechanisms of resistance to Sclerotium rolfsii in peanut (Arachis hypogaea L.) using comparative RNA-Seq analysis of resistant and susceptible genotypes. PLoS ONE 2020, 15, e0236823. [Google Scholar] [CrossRef]
- Dasgupta, U.; Mishra, G.P.; Dikshit, H.K.; Mishra, D.C.; Bosamia, T.; Roy, A.; Bhati, J.; Priti; Aski, M.; Kumar, R.R.; et al. Comparative RNA-Seq analysis unfolds a complex regulatory network imparting yellow mosaic disease resistance in mungbean [Vigna radiata (L.) R. Wilczek]. PLoS ONE 2021, 16, e0244593. [Google Scholar] [CrossRef]
- Sharpe, A.G.; Ramsay, L.; Sanderson, L.-A.; Fedoruk, M.J.; Clarke, W.E.; Li, R.; Kagale, S.; Vijayan, P.; Vandenberg, A.; Bett, K.E. Ancient orphan crop joins modern era: Gene-based SNP discovery and mapping in lentil. BMC Genom. 2013, 14, 192. [Google Scholar] [CrossRef]
- Verma, P.; Shah, N.; Bhatia, S. Development of an expressed gene catalogue and molecular markers rom the de novo assembly of short sequence reads of the lentil (Lens culinaris Medik.) transcriptome. Plant Biotechnol. J. 2013, 11, 894–905. [Google Scholar] [CrossRef]
- Bradley, T.; Moxon, S. FilTar: Using RNA-Seq data to improve microRNA target prediction accuracy in animals. Bioinformatics 2020, 36, 2410–2416. [Google Scholar] [CrossRef]
- Dueck, A.; Ziegler, C.; Eichner, A.; Berezikov, E.; Meister, G. microRNAs associated with the different human Argonaute proteins. Nucleic Acids Res. 2012, 40, 9850–9862. [Google Scholar] [CrossRef]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: MicroRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef]
- McHale, L.; Tan, X.; Koehl, P.; Michelmore, R.W. Plant NBS-LRR proteins: Adaptable guards. Genome Biol. 2006, 7, 212. [Google Scholar] [CrossRef]
- Khorramdelazad, M.; Bar, I.; Whatmore, P.; Smetham, G.; Bhaskarla, V.; Yang, Y.; Bai, S.H.; Mantri, N.; Zhou, Y.; Ford, R. Transcriptome profiling of lentil (Lens culinaris) through the first 24 hours of Ascochyta lentis infection reveals key defence response genes. BMC Genom. 2018, 19, 108. [Google Scholar] [CrossRef]
- Duarte, V.; Salcedo, S.S.; Barreto, R.W. Rhizoctonia solani AG4 causes lentil damping-off in Brazil. Australas. Plant Dis. Notes 2018, 13, 44. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and Direct Sequencing of Fungal Ribosomal RNA Genes for Phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J., White, T.J., Eds.; Academic Press Inc.: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Sharma, A.B.; Kumar, A.; Javeria, S. Pathogenic association of Albifimbria terrestris with rice (Oryzae sativa) seeds. Indian Phytopathol. 2021, 74, 849–850. [Google Scholar] [CrossRef]
- Deepa Sunkad, G.; Sharma, M.; Mallesh, S.B.; Mannur, D.M.; Sreenivas, A.G. Integrated management of dry root rot caused by Rhizoctoniabataticola in chickpea. Int. J. Curr. Microbiol. App. Sci. 2018, 7, 201–209. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualiza-tion and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Smuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Urban, M.; Cuzick, A.; Rutherford, K.; Irvine, A.; Pedro, H.; Pant, R.; Sadanadan, V.; Khamari, L.; Billal, S.; Mohanty, S.; et al. PHI-base: A new interface and further additions for the multi-species pathogen–host interactions database. Nucleic Acids Res. 2017, 45, D604–D610. [Google Scholar] [CrossRef]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; De Silva, N.; Martinez, M.C.; Pedro, H.; Yates, A.D.; et al. PHI-base: The pathogen–host interactions database. Nucleic Acids Res. 2020, 48, D613–D620. [Google Scholar] [CrossRef]
- Numnark, S.; Mhuantong, W.; Ingsriswang, S.; Wichadakul, D. C-mii: A tool for plant miRNA and target identification. BMC Genom. 2012, 13, S16. [Google Scholar] [CrossRef]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Zimmermann, P.; Hirsch-Hoffmann, M.; Hennig, L.; Gruissem, W. Genevestigator. Arabidopsis Microarray Database and Analysis Toolbox. Plant Physiol. 2004, 136, 2621–2632. [Google Scholar] [CrossRef]
- Hruz, T.; Laule, O.; Szabo, G.; Wessendorp, F.; Bleuler, S.; Oertle, L.; Widmayer, P.; Gruissem, W.; Zimmermann, P. Genevestigator V3: A Reference Expression Database for the Meta-Analysis of Transcriptomes. Adv. Bioinform. 2008, 2008, 420747. [Google Scholar] [CrossRef]
- Parmeter, J.R., Jr.; Whitney, H.S. Taxonomy and nomenclature of the imperfect state. In Rhizoctonia solani: Biology and Pathology; University of California Press: Berkeley, CA, USA, 1970; pp. 7–19. [Google Scholar]
- Hosseini, S.Z.; Ismaili, A.; Nazarian-Firouzabadi, F.; Fallahi, H.; Nejad, A.R.; Sohrabi, S.S. Dissecting the molecular responses of lentil to individual and combined drought and heat stresses by comparative transcriptomic analysis. Genomics 2021, 113, 693–705. [Google Scholar] [CrossRef]
- Van Dam, S.; Võsa, U.; Van Der Graaf, A.; Franke, L.; De Magalhães, J.P. Gene co-expression analysis for functional classification and gene–disease predictions. Brief. Bioinform. 2017, 19, 575–592. [Google Scholar] [CrossRef]
- Morris, E.R.; Walker, J.C. Receptor-like protein kinases: The keys to response. Curr. Opin. Plant Biol. 2003, 6, 339–342. [Google Scholar] [CrossRef]
- Afzal, A.J.; Wood, A.J.; Lightfoot, D.A. Plant receptor-like serine threonine kinases: Roles in signaling and plant defense. Mol. Plant Microbe Interact. 2008, 21, 507–517. [Google Scholar] [CrossRef]
- Mengiste, T. Plant Immunity to Necrotrophs. Annu. Rev. Phytopathol. 2012, 50, 267–294. [Google Scholar] [CrossRef]
- Romeis, T.; Ludwig, A.A.; Martin, R.; Jones, J.D.G. Calcium-dependent protein kinases play an essential role in a plant defence response. EMBO J. 2001, 20, 5556–5567. [Google Scholar] [CrossRef]
- Greeff, C.C.; Roux, M.M.; Mundy, J.J.; Petersen, M.M. Receptor-like kinase complexes in plant innate immunity. Front. Plant Sci. 2012, 3, 209. [Google Scholar] [CrossRef] [PubMed]
- Roundhill, S.J.; Fineran, B.A.; Cole, A.L.J.; Ingerfeld, M. Structural aspects of Ascochyta blight of lentil. Can. J. Bot. 1995, 73, 485–497. [Google Scholar] [CrossRef]
- Skłodowska, M.; Gajewska, E.; Kuźniak, E.; Wielanek, M.; Mikiciński, A.; Sobiczewski, P. Antioxidant Profile and Polyphenol Oxidase Activities in Apple Leaves after Erwinia amylovora Infection and Pretreatment with a Benzothiadiazole-type Resistance Inducer (BTH). J. Phytopathol. 2011, 159, 495–504. [Google Scholar] [CrossRef]
- Sambasivam, P.; Taylor, P.W.J.; Ford, R. Pathogenic variation and virulence related responses of Ascochyta lentis on lentil. Eur. J. Plant Pathol. 2017, 147, 265–277. [Google Scholar] [CrossRef]
- Mustafa, B.M.; Coram, T.E.; Pang, E.C.K.; Taylor, P.W.J.; Ford, R. A cDNA microarray approach to decipher lentil (Lens culinaris) responses to Ascochyta lentis. Australas. Plant Pathol. 2009, 38, 617–631. [Google Scholar] [CrossRef]
- Saikia, R.; Singh, B.P.; Kumar, R.; Arora, D.K. Detection of pathogenesis-related proteins-chitinase and β-1,3-glucanase in induced chickpea. Curr. Sci. 2005, 89, 659–663. [Google Scholar]
- Ren, Y.-Y.; West, C.A. Elicitation of Diterpene Biosynthesis in Rice (Oryza sativa L.) by Chitin. Plant Physiol. 1992, 99, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Vaghefi, N.; Mustafa, B.; Dulal, N.; Selby-Pham, J.; Taylor, P.; Ford, R. A novel pathogenesis-related protein (LcPR4a) from lentil, and its involvement in defence against Ascochyta lentis. Phytopathol. Mediterr. 2013, 52, 192–201. [Google Scholar]
- Liu, J.-J.; Ekramoddoullah, A.K. The family 10 of plant pathogenesis-related proteins: Their structure, regulation, and function in response to biotic and abiotic stresses. Physiol. Mol. Plant Pathol. 2006, 68, 3–13. [Google Scholar] [CrossRef]
- Raiola, A.; Lionetti, V.; Elmaghraby, I.; Immerzeel, P.; Mellerowicz, E.J.; Salvi, G.; Cervone, F.; Bellincampi, D. Pectin Methylesterase Is Induced in Arabidopsis upon Infection and Is Necessary for a Successful Colonization by Necrotrophic Pathogens. Mol. Plant-Microbe Interact. 2011, 24, 432–440. [Google Scholar] [CrossRef] [PubMed]
- De Lorenzo, G.; D’Ovidio, R.; Cervone, F. The role of polygalacturonase-inhibiting proteins (pgips) in defense against pathogenic fungi. Annu. Rev. Phytopathol. 2001, 39, 313–335. [Google Scholar] [CrossRef] [PubMed]
- Ghanashyam, C.; Jain, M. Role of auxin-responsive genes in biotic stress responses. Plant Signal. Behav. 2009, 4, 846–848. [Google Scholar] [CrossRef]
- Van Ooijen, G.; Burg, H.A.V.D.; Cornelissen, B.J.C.; Takken, F.L.W. Structure and Function of Resistance Proteins in Solanaceous Plants. Annu. Rev. Phytopathol. 2007, 45, 43–72. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Tian, Z.; Liu, J.; Song, B.; Xie, C. Cloning and molecular characterization of the potato RING finger protein gene StRFP1 and its function in potato broad-spectrum resistance against Phytophthora infestans. J. Plant Physiol. 2010, 167, 488–496. [Google Scholar] [CrossRef]
- Fu, Z.Q.; Dong, X. Systemic acquired resistance: Turning local infection into global defense. Annu. Rev. Plant Biol. 2013, 64, 839–863. [Google Scholar] [CrossRef]
- Achard, P.; Renou, J.-P.; Berthomé, R.; Harberd, N.P.; Genschik, P. Plant DELLAs Restrain Growth and Promote Survival of Adversity by Reducing the Levels of Reactive Oxygen Species. Curr. Biol. 2008, 18, 656–660. [Google Scholar] [CrossRef]
- Vidhyasekaran, P. Molecular manipulation of transcription factors, the master regulators of PAMP-triggered signaling systems. In Switching on Plant Innate Immunity Signaling Systems; Springer: Berlin/Heidelberg, Germany, 2016; pp. 255–358. [Google Scholar]
- Ludwig, A.A.; Saitoh, H.; Felix, G.; Freymark, G.; Miersch, O.; Wasternack, C.; Boller, T.; Jones, J.D.G.; Romeis, T. Ethylene-mediated cross-talk between calcium-dependent protein kinase and MAPK signaling controls stress responses in plants. Proc. Natl. Acad. Sci. USA 2005, 102, 10736–10741. [Google Scholar] [CrossRef]
- Han, Y.; Zhang, K.; Yang, J.; Zhang, N.; Fang, A.; Zhang, Y.; Liu, Y.; Chen, Z.; Hsiang, T.; Sun, W. Differential expression profiling of the early response to Ustilaginoidea virens between false smut resistant and susceptible rice varieties. BMC Genom. 2015, 16, 955. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, L.; Li, X.; Zhang, Y.; Xu, S.; Huang, X. Prediction of miRNA targets by learning from interaction sequences. PLoS ONE 2020, 15, e0232578. [Google Scholar] [CrossRef] [PubMed]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Wessels, H.-H.; Lebedeva, S.; Hirsekorn, A.; Wurmus, R.; Akalin, A.; Mukherjee, N.; Ohler, U. Global identification of functional microRNA-mRNA interactions in Drosophila. Nat. Commun. 2019, 10, 1626. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Chu, L.; Jin, Q.; Wang, Y.; Chen, X.; Zhao, H.; Xue, Z. Transcriptome-Wide Identification of miRNAs and Their Targets from Typha angustifolia by RNA-Seq and Their Response to Cadmium Stress. PLoS ONE 2015, 10, e0125462. [Google Scholar] [CrossRef]
- Yin, X.; Wang, J.; Cheng, H.; Wang, X.; Yu, D. Detection and evolutionary analysis of soybean miRNAs responsive to soybean mosaic virus. Planta 2013, 237, 1213–1225. [Google Scholar] [CrossRef]
- Zheng, C.; Ye, M.; Sang, M.; Wu, R. A Regulatory Network for miR156-SPL Module in Arabidopsis thaliana. Int. J. Mol. Sci. 2019, 20, 6166. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Zhang, S.; Liu, D.; Duan, Y.; Dong, W. Identification of Soybean MicroRNAs Involved in Soybean Cyst Nematode Infection by Deep Sequencing. PLoS ONE 2012, 7, e39650. [Google Scholar] [CrossRef]
- Ramesh, S.V.; Chouhan, B.; Kumar, G.; Praveen, S.; Chand, S. Expression dynamics of Glycine max (L.) Merrill microRNAs (miRNAs) and their targets during Mungbean yellow mosaic India virus (MYMIV) infection. Physiol. Mol. Plant Pathol. 2017, 100, 13–22. [Google Scholar] [CrossRef]
- Kulcheski, F.R.; De Oliveira, L.F.; Molina, L.G.; Almerão, M.P.; Rodrigues, F.A.; Marcolino, J.; Barbosa, J.F.; Stolf-Moreira, R.; Nepomuceno, A.L.; Marcelino-Guimarães, F.C.; et al. Identification of novel soybean microRNAs involved in abiotic and biotic stresses. BMC Genom. 2011, 12, 307–317. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Sample Name | |||
|---|---|---|---|---|
| Inoculated (Root-A1) | Inoculated (Root-A2) | Control (Root-B1) | Control (Root-B2) | |
| Read length | 100 × 2 | 100 × 2 | 100 × 2 | 100 × 2 |
| Raw reads | 110,534,542 | 107,467,648 | 119,708,340 | 114,381,960 |
| Clean read | 110,235,896 | 107,170,090 | 119,341,370 | 113,806,616 |
| Clean bases (Mb) | 10,981.28 | 10,662.28 | 11,879.05 | 11,343.24 |
| GC contents (%) | 43.07 | 42.88 | 44.01 | 43.95 |
| Q30 (%) | 93.64 | 94.23 | 93.84 | 94.12 |
| Total No. of paired end (PE) reads | 55,117,948 | 53,585,045 | 59,670,685 | 56,903,308 |
| Read alignment (%) | 96.82 | 97.06 | 96.93 | 97.05 |
| Unigenes (No) with FPKM ≥ 1 | 37,242 | 37,631 | 36,421 | 37,419 |
| Description/Annotation Category | Number of Unigenes |
|---|---|
| Total number of Unigenes considered for BLASTX | 42,606 |
| Number of Unigenes with significant BLASTX match | 38,437 |
| Number of Unigenes with a significant BLASTX hit against (Rhizoctonia protein sequences) | 12,648 |
| Number of Unigenes with a significant BLASTX hit against (Uniprot plant protein database) | 25,789 |
| Number of unigenes with No Blastx hit | 4169 |
| Categories | Count | Percentage |
|---|---|---|
| Reduced virulence | 331 | 45.66 |
| Unaffected pathogenicity | 208 | 28.69 |
| Loss of pathogenicity | 92 | 12.69 |
| lethal | 47 | 6.48 |
| Increased virulence (hypervirulence) | 23 | 3.17 |
| Effector (plant avirulence determinant) | 17 | 2.34 |
| Resistance to chemical | 4 | 0.55 |
| Sensitivity to chemical | 3 | 0.41 |
| Total | 725 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, G.P.; Aski, M.S.; Bosamia, T.; Chaurasia, S.; Mishra, D.C.; Bhati, J.; Kumar, A.; Javeria, S.; Tripathi, K.; Kohli, M.; et al. Insights into the Host-Pathogen Interaction Pathways through RNA-Seq Analysis of Lens culinaris Medik. in Response to Rhizoctonia bataticola Infection. Genes 2022, 13, 90. https://doi.org/10.3390/genes13010090
Mishra GP, Aski MS, Bosamia T, Chaurasia S, Mishra DC, Bhati J, Kumar A, Javeria S, Tripathi K, Kohli M, et al. Insights into the Host-Pathogen Interaction Pathways through RNA-Seq Analysis of Lens culinaris Medik. in Response to Rhizoctonia bataticola Infection. Genes. 2022; 13(1):90. https://doi.org/10.3390/genes13010090
Chicago/Turabian StyleMishra, Gyan P., Muraleedhar S. Aski, Tejas Bosamia, Shiksha Chaurasia, Dwijesh Chandra Mishra, Jyotika Bhati, Atul Kumar, Shaily Javeria, Kuldeep Tripathi, Manju Kohli, and et al. 2022. "Insights into the Host-Pathogen Interaction Pathways through RNA-Seq Analysis of Lens culinaris Medik. in Response to Rhizoctonia bataticola Infection" Genes 13, no. 1: 90. https://doi.org/10.3390/genes13010090
APA StyleMishra, G. P., Aski, M. S., Bosamia, T., Chaurasia, S., Mishra, D. C., Bhati, J., Kumar, A., Javeria, S., Tripathi, K., Kohli, M., Kumar, R. R., Singh, A. K., Devi, J., Kumar, S., & Dikshit, H. K. (2022). Insights into the Host-Pathogen Interaction Pathways through RNA-Seq Analysis of Lens culinaris Medik. in Response to Rhizoctonia bataticola Infection. Genes, 13(1), 90. https://doi.org/10.3390/genes13010090