
Improved High-Quality Genome Assembly and Annotation of Pineapple (Ananas comosus) Cultivar MD2 Revealed Extensive Haplotype Diversity and Diversified FRS/FRF Gene Family

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material Collection, DNA and RNA Extraction, and Sequencing
2.2. Genome Assembly, Phasing, and Scaffolding
2.3. Assembly Error Correction and Gap-Filling
2.4. Genome Quality Analysis
2.5. Genome Annotation
2.6. Haplotype Comparison
2.7. Identification and Characterization of FRS/FRF Transcription Factors
3. Results and Discussion
3.1. Genome Assembly and Quality Assessment
3.2. Genome Assembly Comparisons
3.3. Repetitive Sequence Annotation and Analysis
3.4. Gene Prediction and Annotation
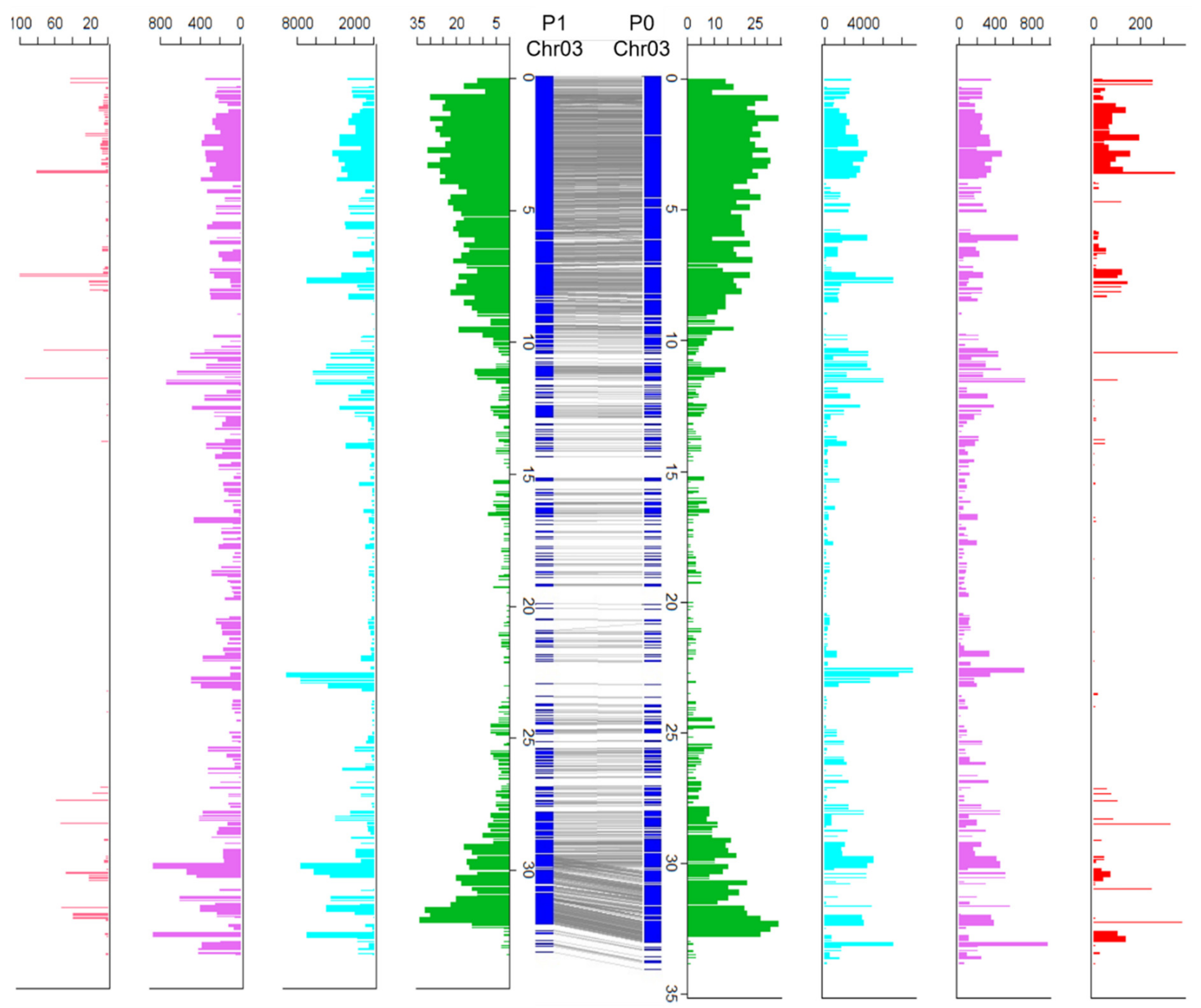
3.5. Haplotype Comparison
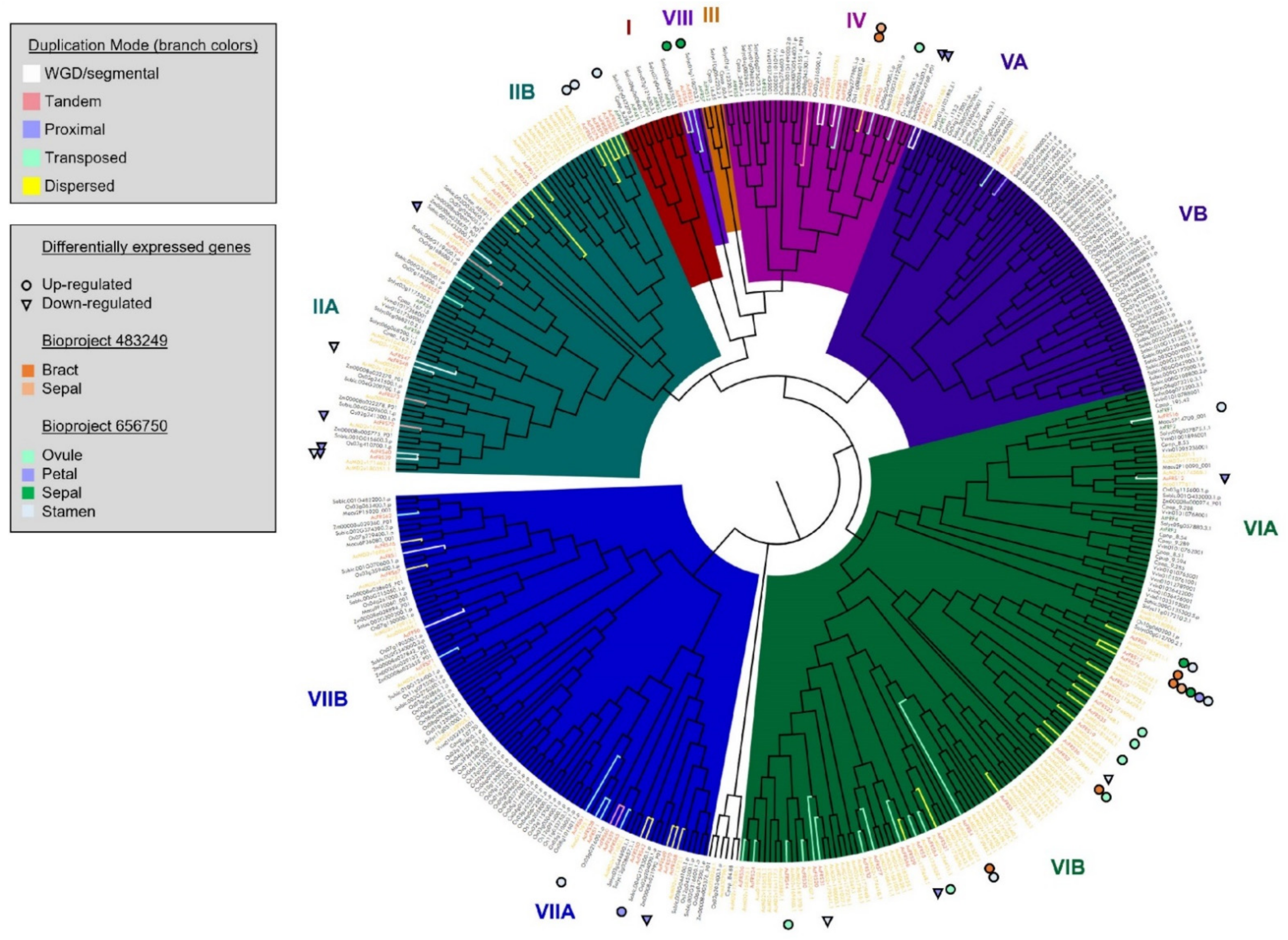
3.6. Characterization of FRS/FRF Family Transcription Factors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wali, N. Chapter 3.34—Pineapple (Ananas comosus). In Nonvitamin and Nonmineral Nutritional Supplements; Nabavi, S.M., Silva, A.S., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 367–373. ISBN 978-0-12-812491-8. [Google Scholar]
- Redwan, R.M.; Saidin, A.; Kumar, S.V. The draft genome of MD-2 pineapple using hybrid error correction of long reads. DNA Res. 2016, 23, 427–439. [Google Scholar] [CrossRef]
- Ahmar, S.; Gill, R.A.; Ki-Hong, J.; Faheem, A.; Qasim, M.U.; Mubeen, M.; Zhou, W. Conventional and Molecular Techniques from Simple Breeding to Speed Breeding in Crop Plants: Recent Advances and Future Outlook. Int. J. Mol. Sci. 2020, 21, 2590. [Google Scholar] [CrossRef] [Green Version]
- Abdul Rahman, A.; Kumar, V. Estimation of the Pineapple Genome Size by Using Quantitative Real-Time Polymerase Chain Reaction. In Proceedings of the 9th Malaysia Genetics Congress, 9th Malaysia Genetics Congress, Kuching, Malaysia, 28–30 September 2011. [Google Scholar]
- Ming, R.; VanBuren, R.; Wai, C.M.; Tang, H.; Schatz, M.C.; Bowers, J.E.; Lyons, E.; Wang, M.-L.; Chen, J.; Biggers, E.; et al. The pineapple genome and the evolution of CAM photosynthesis. Nat. Genet. 2015, 47, 1435–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Lin, F.; An, D.; Wang, W.; Huang, R. Genome Sequencing and Assembly by Long Reads in Plants. Genes 2017, 9, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kronenberg, Z.N.; Hall, R.J.; Hiendleder, S.; Smith, T.P.L.; Sullivan, S.T.; Williams, J.L.; Kingan, S.B. FALCON-Phase: Integrating PacBio and Hi-C data for phased diploid genomes. bioRxiv 2018, 327064. [Google Scholar] [CrossRef] [Green Version]
- Lajoie, B.R.; Dekker, J.; Kaplan, N. The Hitchhiker’s Guide to Hi-C Analysis: Practical guidelines. Methods 2015, 72, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Kumar, V.; Olson, A.; Ware, D. Reviving the Transcriptome Studies: An Insight Into the Emergence of Single-Molecule Transcriptome Sequencing. Front. Genet. 2019, 10, 384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Li, G. FAR1-RELATED SEQUENCE (FRS) and FRS-RELATED FACTOR (FRF) Family Proteins in Arabidopsis Growth and Development. Front. Plant. Sci. 2018, 9, 692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Ma, M.; Li, G.; Yuan, L.; Xie, Y.; Wei, H.; Ma, X.; Li, Q.; Devlin, P.F.; Xu, X.; et al. Transcription Factors FHY3 and FAR1 Regulate Light-Induced CIRCADIAN CLOCK ASSOCIATED1 Gene Expression in Arabidopsis. Plant. Cell 2020, 32, 1464–1478. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Ding, L.; Casola, C.; Ripoll, D.R.; Feschotte, C.; Wang, H. Transposase-derived transcription factors regulate light signaling in Arabidopsis. Science 2007, 318, 1302–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.; Wang, W.; Chen, D.; Ji, Q.; Jing, Y.; Wang, H.; Lin, R. Transposase-derived proteins FHY3/FAR1 interact with PHYTOCHROME-INTERACTING FACTOR1 to regulate chlorophyll biosynthesis by modulating HEMB1 during deetiolation in Arabidopsis. Plant. Cell 2012, 24, 1984–2000. [Google Scholar] [CrossRef] [Green Version]
- Charlotte, A.O.A.; Gbènato, A.-D.E.; Clément, A. Optimizing Genomic DNA Isolation in Pineapple (Ananas comosus L.). J. Plant. Breed. Genet. 2016, 4, 11–18. [Google Scholar]
- Chin, C.-S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O’Malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A.; et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faust, G.G.; Hall, I.M. SAMBLASTER: Fast duplicate marking and structural variant read extraction. Bioinformatics 2014, 30, 2503–2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.-Y.; VanBuren, R.; Paris, M.; Zhou, H.; Zhang, X.; Wai, C.M.; Yan, H.; Chen, S.; Alonge, M.; Ramakrishnan, S.; et al. The bracteatus pineapple genome and domestication of clonally propagated crops. Nat. Genet. 2019, 51, 1549–1558. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Yu, Q.; Shi, Y.; Hua, X.; Tang, H.; Yang, L.; Ming, R.; Zhang, J. PGD: Pineapple Genomics Database. Hortic. Res. 2018, 5, 66. [Google Scholar] [CrossRef] [Green Version]
- Durand, N.C.; Robinson, J.T.; Shamim, M.S.; Machol, I.; Mesirov, J.P.; Lander, E.S.; Aiden, E.L. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 2016, 3, 99–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLOS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.-C.; Xu, T.-J.; Zhu, R.; Zhang, Y.; Li, S.-Q.; Wang, H.-W.; Li, J.-T. LR_Gapcloser: A tiling path-based gap closer that uses long reads to complete genome assembly. Gigascience 2019, 8, giy157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, S.; Chen, J.; Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 2018, 46, e126. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2017, 35, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [Green Version]
- Keilwagen, J.; Hartung, F.; Paulini, M.; Twardziok, S.O.; Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinform. 2018, 19, 189. [Google Scholar] [CrossRef] [Green Version]
- Keilwagen, J.; Wenk, M.; Erickson, J.L.; Schattat, M.H.; Grau, J.; Hartung, F. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016, 44, e89. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.-Y.; Krishnakumar, V.; Chan, A.P.; Thibaud-Nissen, F.; Schobel, S.; Town, C.D. Araport11: A complete reannotation of the Arabidopsis thaliana reference genome. Plant. J. 2017, 89, 789–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ming, R.; Hou, S.; Feng, Y.; Yu, Q.; Dionne-Laporte, A.; Saw, J.H.; Senin, P.; Wang, W.; Ly, B.V.; Lewis, K.L.T.; et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 2008, 452, 991–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Droc, G.; Larivière, D.; Guignon, V.; Yahiaoui, N.; This, D.; Garsmeur, O.; Dereeper, A.; Hamelin, C.; Argout, X.; Dufayard, J.-F.; et al. The banana genome hub. Database 2013, 2013, bat035. [Google Scholar] [CrossRef]
- Jain, R.; Jenkins, J.; Shu, S.; Chern, M.; Martin, J.A.; Copetti, D.; Duong, P.Q.; Pham, N.T.; Kudrna, D.A.; Talag, J.; et al. Genome sequence of the model rice variety KitaakeX. BMC Genom. 2019, 20, 905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, X.; Yang, J.; Sun, S.; Yang, W. Identification and Analysis of Resistance-like Genes in the Tomato Genome. J. Phytopathol. 2014, 162, 137–146. [Google Scholar] [CrossRef]
- McCormick, R.F.; Truong, S.K.; Sreedasyam, A.; Jenkins, J.; Shu, S.; Sims, D.; Kennedy, M.; Amirebrahimi, M.; Weers, B.D.; McKinley, B.; et al. The Sorghum bicolor reference genome: Improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. Plant. J. 2018, 93, 338–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaillon, O.; Aury, J.-M.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, C.N.; Hirsch, C.D.; Brohammer, A.B.; Bowman, M.J.; Soifer, I.; Barad, O.; Shem-Tov, D.; Baruch, K.; Lu, F.; Hernandez, A.G.; et al. Draft Assembly of Elite Inbred Line PH207 Provides Insights into Genomic and Transcriptome Diversity in Maize. Plant. Cell 2016, 28, 2700–2714. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Sinharoy, S.; Udvardi, M.; Zhao, P.X. PlantTFcat: An online plant transcription factor and transcriptional regulator categorization and analysis tool. BMC Bioinform. 2013, 14, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osuna-Cruz, C.M.; Paytuvi-Gallart, A.; Di Donato, A.; Sundesha, V.; Andolfo, G.; Aiese Cigliano, R.; Sanseverino, W.; Ercolano, M.R. PRGdb 3.0: A comprehensive platform for prediction and analysis of plant disease resistance genes. Nucleic Acids Res. 2018, 46, D1197–D1201. [Google Scholar] [CrossRef]
- Ou, S.; Su, W.; Liao, Y.; Chougule, K.; Agda, J.R.A.; Hellinga, A.J.; Lugo, C.S.B.; Elliott, T.A.; Ware, D.; Peterson, T.; et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 2019, 20, 275. [Google Scholar] [CrossRef] [Green Version]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 9, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, S.; Jiang, N. LTR_FINDER_parallel: Parallelization of LTR_FINDER enabling rapid identification of long terminal repeat retrotransposons. Mob. DNA 2019, 10, 48. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.; Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant. Physiol. 2018, 176, 1410–1422. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Liang, C. Generic Repeat Finder: A High-Sensitivity Tool for Genome-Wide De Novo Repeat Detection. Plant. Physiol. 2019, 180, 1803–1815. [Google Scholar] [CrossRef]
- Su, W.; Gu, X.; Peterson, T. TIR-Learner, a New Ensemble Method for TIR Transposable Element Annotation, Provides Evidence for Abundant New Transposable Elements in the Maize Genome. Mol. Plant. 2019, 12, 447–460. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.; He, L.; Lai, J.; Dooner, H.K.; Du, C. HelitronScanner uncovers a large overlooked cache of Helitron transposons in many plant genomes. Proc. Natl. Acad. Sci. USA 2014, 111, 10263–10268. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Tang, D.; Huang, W.; Yang, Z.; Zhang, Y.; Hamilton, J.P.; Visser, R.G.F.; Bachem, C.W.B.; Robin Buell, C.; Zhang, Z.; et al. Haplotype-resolved genome analyses of a heterozygous diploid potato. Nat. Genet. 2020, 52, 1018–1023. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Fischer, S.; Brunk, B.P.; Chen, F.; Gao, X.; Harb, O.S.; Iodice, J.B.; Shanmugam, D.; Roos, D.S.; Stoeckert, C., Jr. J. Using OrthoMCL to Assign Proteins to OrthoMCL-DB Groups or to Cluster Proteomes Into New Ortholog Groups. Curr. Protoc. Bioinform. 2011, 35, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Ma, T.; Kang, M.; Ai, F.; Zhang, J.; Dong, G.; Liu, J. A high-quality Actinidia chinensis (kiwifruit) genome. Hortic. Res. 2019, 6, 117. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Li, Y.; Jin, X.; Liu, L.; Dai, X.; Liu, Y.; Zhao, L.; Zheng, P.; Wang, X.; Liu, Y.; et al. Floral transcriptomes reveal gene networks in pineapple floral growth and fruit development. Commun. Biol. 2020, 3, 500. [Google Scholar] [CrossRef]
- Liu, S.; Yang, L.; Li, J.; Tang, W.; Li, J.; Lin, R. FHY3 interacts with phytochrome B and regulates seed dormancy and germination. Plant. Physiol. 2021, 187, 289–302. [Google Scholar] [CrossRef]
- Ritter, A.; Iñigo, S.; Fernández-Calvo, P.; Heyndrickx, K.S.; Dhondt, S.; Shi, H.; De Milde, L.; Vanden Bossche, R.; De Clercq, R.; Eeckhout, D.; et al. The transcriptional repressor complex FRS7-FRS12 regulates flowering time and growth in Arabidopsis. Nat. Commun. 2017, 8, 15235. [Google Scholar] [CrossRef]
- Fernández-Calvo, P.; Iñigo, S.; Glauser, G.; Vanden Bossche, R.; Tang, M.; Li, B.; De Clercq, R.; Nagels Durand, A.; Eeckhout, D.; Gevaert, K.; et al. FRS7 and FRS12 recruit NINJA to regulate expression of glucosinolate biosynthesis genes. New Phytol. 2020, 227, 1124–1137. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Wang, H. Arabidopsis FHY3/FAR1 gene family and distinct roles of its members in light control of Arabidopsis development. Plant. Physiol. 2004, 136, 4010–4022. [Google Scholar] [CrossRef] [Green Version]
- Qiao, X.; Li, Q.; Yin, H.; Qi, K.; Li, L.; Wang, R.; Zhang, S.; Paterson, A.H. Gene duplication and evolution in recurring polyploidization–diploidization cycles in plants. Genome Biol. 2019, 20, 38. [Google Scholar] [CrossRef] [Green Version]
- Zhong, M.; Jiang, X.-D.; Weihua, C.; Hu, J.-Y. Expansion and expression diversity of FAR1/FRS-like genes provides insights into flowering time regulation in roses. Plant. Divers. 2020, 43, 173–179. [Google Scholar] [CrossRef]
- Sharma, A.; Wai, C.M.; Ming, R.; Yu, Q. Diurnal Cycling Transcription Factors of Pineapple Revealed by Genome-Wide Annotation and Global Transcriptomic Analysis. Genome Biol. Evol. 2017, 9, 2170–2190. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MD2 v2 P0 | MD2 v2 P1 | |||||
|---|---|---|---|---|---|---|
| # | Length [bp] | % | # | Length [bp] | % | |
| Assembly feature | ||||||
| Sequences | 63 | 543,505,080 | 96.5 */101.0 ** | 63 | 531,615,398 | 94.4 */98.8 ** |
| Contigs | 812 | 543,433,016 | 96.5 */101.0 ** | 805 | 531,544,088 | 94.4 */98.8 ** |
| Max. sequence length | 43,498,842 | 42,511,760 | ||||
| Min. contig length | 5678 | 4092 | ||||
| Max. contig length | 5,969,083 | 5,971,173 | ||||
| Contig N50 length | 1,524,720 | 1,521,169 | ||||
| Scaffold N50 length | 21,996,178 | 23,016,244 | ||||
| Chromosome-anchored sequence | 541,772,120 | 99.7 | 529,885,665 | 99.7 | ||
| Genome annotation | ||||||
| Transposable element content | 337,179,261 | 62.0 | 329,341,567 | 62.0 | ||
| Gene models | 30,591 | 35,382,141 | 29,550 | 34,906,836 | ||
| Genes in pseudomolecules | 30,590 | 35,382,088 | 100 | 29,550 | 34,906,836 | 100 |
| MD2 v2 P0 vs. MD2 v1 | MD2 v2 P0 vs. F153 v7 | MD2 v2 P0 vs. CB5 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| # | Length [bp] | %/Fold Change | # | Length [bp] | %/Fold Change | # | Length [bp] | %/Fold Change | |
| Assembly feature | |||||||||
| Sequences | −8385 | +19,435,418 | +3.5% */+3.6% ** | −3070 | +161,599,861 | +28.7% */+30.7% ** | −40 | +30,269,689 | +5.4% */+5.6% ** |
| Contigs | −20,927 | +33,521,821 | +6.0% */+6.2% ** | −8550 | +168,298,756 | +29.9% */32.0% ** | −1158 | +30,384,325 | +5.4% */+5.6% ** |
| Max. sequence length | +42,211,785 | +33.8 Fold | +26,164,174 | +2.5 Fold | +16,366,087 | +1.6 Fold | |||
| Min. contig length | +5677 | +5678 Fold | +5497 | +30.4 Fold | −6456 | −2.1 Fold | |||
| Max. contig length | +4,742,061 | +4.9 Fold | +4,957,247 | +5.9 Fold | +3,781,837 | +2.7 Fold | |||
| Contig N50 length | +1,467,419 | +26.6 Fold | +1,410,093 | +13.3 Fold | +1,098,024 | +3.6 Fold | |||
| Scaffold N50 length | +21,843,094 | +143.7 Fold | +10,290,687 | +1.9 Fold | +1,817,735 | +1.1 Fold | |||
| Chromosome-anchored sequence * | +541,772,120 | NA | +225,928,334 | +40.1% | +55,160,525 | +9.8% | |||
| Genome annotation | |||||||||
| Transposable element content | +26,531,143 | +2.76% | +136,080,198 | +10.03% | +38,062,886 | +3.76% | |||
| Gene models | +6993 | +7,217,109 | +3567 | +3,729,585 | NA | NA | NA | ||
| Genes in pseudomolecules | NA | NA | +6698 | +6,439,201 | NA | NA | NA | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yow, A.G.; Bostan, H.; Castanera, R.; Ruggieri, V.; Mengist, M.F.; Curaba, J.; Young, R.; Gillitt, N.; Iorizzo, M. Improved High-Quality Genome Assembly and Annotation of Pineapple (Ananas comosus) Cultivar MD2 Revealed Extensive Haplotype Diversity and Diversified FRS/FRF Gene Family. Genes 2022, 13, 52. https://doi.org/10.3390/genes13010052
Yow AG, Bostan H, Castanera R, Ruggieri V, Mengist MF, Curaba J, Young R, Gillitt N, Iorizzo M. Improved High-Quality Genome Assembly and Annotation of Pineapple (Ananas comosus) Cultivar MD2 Revealed Extensive Haplotype Diversity and Diversified FRS/FRF Gene Family. Genes. 2022; 13(1):52. https://doi.org/10.3390/genes13010052
Chicago/Turabian StyleYow, Ashley G., Hamed Bostan, Raúl Castanera, Valentino Ruggieri, Molla F. Mengist, Julien Curaba, Roberto Young, Nicholas Gillitt, and Massimo Iorizzo. 2022. "Improved High-Quality Genome Assembly and Annotation of Pineapple (Ananas comosus) Cultivar MD2 Revealed Extensive Haplotype Diversity and Diversified FRS/FRF Gene Family" Genes 13, no. 1: 52. https://doi.org/10.3390/genes13010052
APA StyleYow, A. G., Bostan, H., Castanera, R., Ruggieri, V., Mengist, M. F., Curaba, J., Young, R., Gillitt, N., & Iorizzo, M. (2022). Improved High-Quality Genome Assembly and Annotation of Pineapple (Ananas comosus) Cultivar MD2 Revealed Extensive Haplotype Diversity and Diversified FRS/FRF Gene Family. Genes, 13(1), 52. https://doi.org/10.3390/genes13010052