1. Introduction
The 1000 Genomes Project (1000G) [
1] corresponds to the first attempt to characterize the worldwide genetic variation in humans. The project was created to generate accurate haplotype information across different human populations. To do so, the project aimed to characterize over 95% of variants in genomic regions that have >1% allele frequency in each of the major population groups (populations in or with ancestry from Europe, East Asia, South Asia, West Africa, and the Americas). The project started with 15 populations in the pilot phase, and had a total of 26 populations by the end of Phase 3, when the project was concluded. In the pilot phase, the project characterized a total of 1092 samples (not evenly distributed across the different populations) and, by the end of the Phase 3, it included a total of 2504 samples (with a close to even distribution across all populations). The final genomic dataset is heterogeneous in nature, comprising individuals with low coverage and exome sequencing data, produced in nine sequencing centers using five sequencing technologies and bioinformatic pipelines. An additional problem is that populations were not divided evenly across all sequencing centers. For example, for the GWD (Gambian in Western Division, Mandinka) population, the BGI sequenced 25 individuals, the Broad Institute sequenced 86 individuals, and Washington University sequenced two individuals. Furthermore, not all centers did all the types of sequencing technologies. For example, the Broad Institute ran low coverage whole genome sequencing and whole exome sequencing. In contrast, the Max Planck Institute for Molecular Genetics only conducted low coverage whole genome sequencing. Furthermore, each center followed its own set of protocols to prepare the samples.
1000G has been one of the cornerstones of population genetics, as it provided the first dataset that considered human worldwide variation. Since then, it has been one of the most widely used human population genetic, medical genetics, and genetic epidemiology datasets. In human population genetics, the 1000G has been widely used for understanding mutation patterns [
2], characterizing the genetic variation of the considered human populations [
3], identifying segments of archaic introgression [
4], studying signatures of positive selection [
5,
6] or as a gold standard for comparing the burden of loss of function variants (LoF) [
3]. In genetic epidemiology, 1000G is routinely used for data phasing and imputation [
7,
8] to increase single nucleotide variant (SNV) density panels genotyped with microarrays. The prevalence of loss of function (LoF) variants in healthy individuals [
9], insights into cancer genomics [
10], short tandem repeats variation [
11], evolution and functional impact of short indels [
12], and many of the results from this project have boosted our understanding of the genomics of the species and the general patterns of diversity present in human populations. Its usage in combination with other whole genome sequencing (WGS) datasets as a reference dataset is also a common practice in the field of medical genomics [
3].
However, some problems related to the genotypes called in 1000G have been already reported. For example, Ref. [
13] could not reproduce a particular mutation signature (*AC →*CC) reported in the 1000G JPT population (Japanese in Tokyo, Japan) by [
14] using a different cohort from the same population (the Nagahama cohort). Furthermore, Ref. [
15] evaluated the accuracy of the phasing in the Phase 3 samples and concluded that the 1000G data are best used to impute common variants (minor allele frequency (MAF) ≥0.01) and has limited utility for imputing rare variants. Finally, Ref. [
16] described sets of SNVs showing patterns of linkage disequilibrium likely due to the presence of sequencing errors, and directly linked them to the sequencing center where the individual was sequenced.
Singletons can be artificially generated in a variety of ways: the sequencer can misread the base that is being incorporated, the base-calling algorithm may not call the proper base, the alignment algorithm may match the read to an incorrect place, or the SNP-calling or the genotyping algorithm may call an SNV where there is none (or vice versa). In order to solve these problems, a series of different solutions have been proposed such as filtering SNVs by means of a quality score (an associated measure of uncertainty to the SNV) [
17], or examining allele distributions across individuals and calculating their fit to an expected distribution [
18].
In this study, we analyzed to what extent these batch effects due to the sequencing center could affect measures of genetic variation related to variants at low frequency, looking at rare events that have been previously used in estimating LoF mutations [
2], patterns of singletons, and archaic introgression. LoF and particularly dominant mutations are associated with Mendelian diseases [
19]. Due to their deleterious nature, they are maintained in general population at a low frequency, except when occurring on dispensable genes or under positive selection [
20]. Therefore, being able to distinguish true loss of function mutations from sequencing artefacts is important for properly assessing the genetic risk of an individual regarding a possible medical condition [
2]. Singletons are the most common genetic variants in the human genome, reflecting the recent demographic history of human populations [
21]. The presence of new mutations can be used for estimating the mutation rate and the environmental and intrinsic factors shaping it [
22]. Finally, a depletion of archaic introgression in the human genome, particularly in functional regions, has been documented. This observation is most likely due to purifying selection against the hybrid [
23]. However, currently introgressed fragments can have a phenotypic impact (e.g., COVID-19 susceptibility [
24]). Therefore, identifying these fragments can be important for understanding the history of archaic introgression, identifying archaic species from which ancient DNA is not yet available [
4,
25], assessing their role in phenotypes of medical interest.
Furthermore, we wanted to assess to what extent sequencing errors in the 1000G could affect the interpretation of results in newly generated data, especially for populations that due to their rural condition could be potential isolates. As a proof of concept, we used the rural population of the Spanish Eastern Pyrenees (SEP), presented and analyzed in [
26].
4. Discussion
The finalization of the 1000G project represented a milestone for the human population genetics community [
1]. This dataset is normally used as basis for imputation in microarrays [
48], to phase genomes [
7] in evolutionary studies [
2,
4,
5,
49], in multiple medical studies [
50], or as a basis to identify potential genetic isolates [
3].
However, two studies [
13,
15] have recently raised concerns about the presence of batch effects at variants at low frequency. The study [
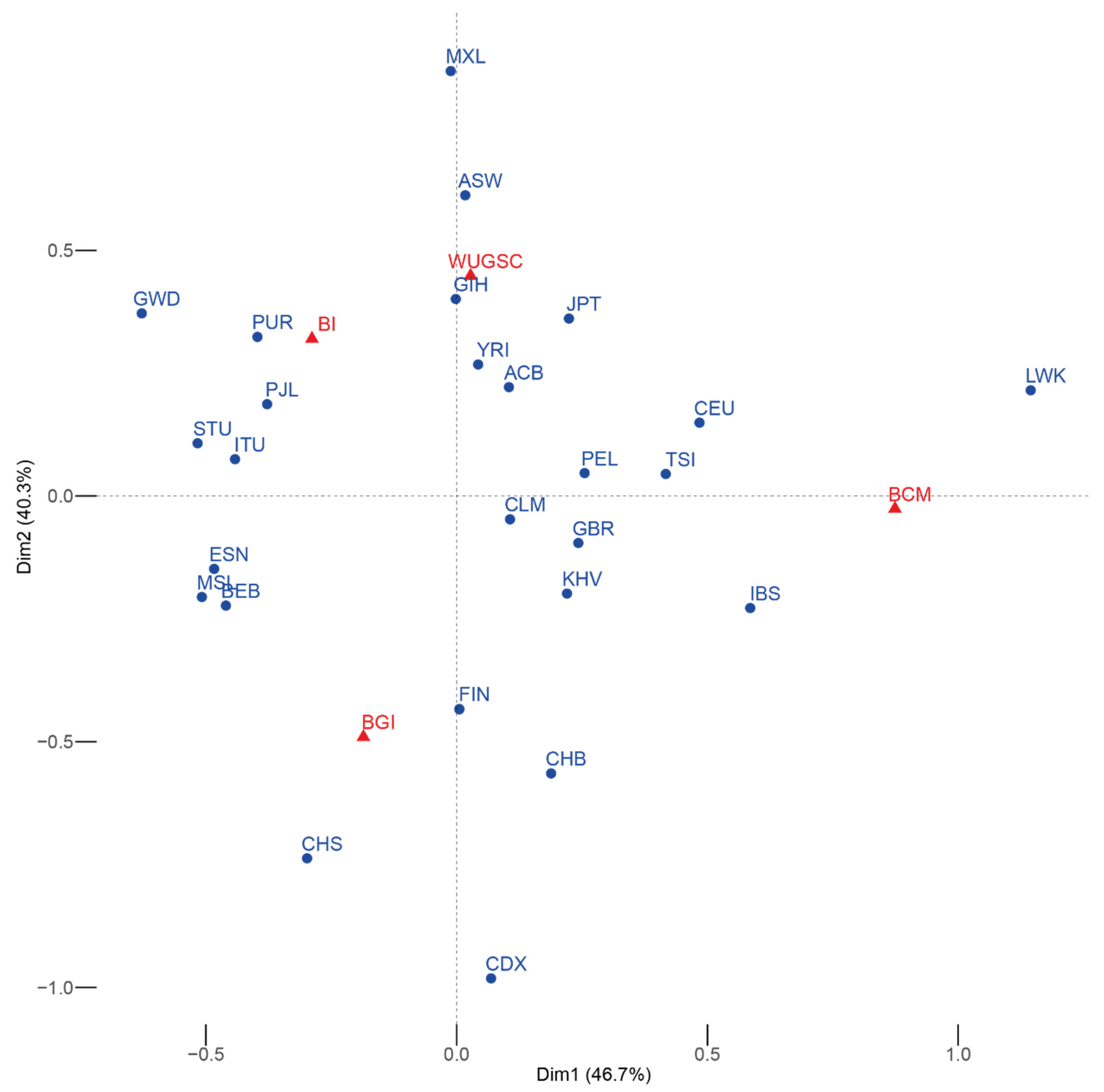
15] in particular pointed to the sequencing center as one of the main factors affecting the presence of rare spurious mutations in 1000G individuals. Given these results, in this study we looked at to what extent the sequencing center, as reported by the spreadsheet of the 1000G (
https://www.internationalgenome.org/data/ accessed on 26 April 2019), could influence statistics of population genomics that quantify variants at low frequency in the human genome. Across this paper we have presented proof that some variation present in the 1000 Genomes Project dataset seems to have a noticeable batch effect regarding the sequencing center of the sample, although this batch effect is not noticeable for common variants (MAF > 5%).
LoF variants are usually present at low frequency in the population due to their deleterious effects in carriers and their consequent erasure from the population by purifying selection [
51]. Given the fact that 1000G individuals are healthy [
1], the presence of LoF variants is expected in heterozygosis and to be mostly recessively inherited. Therefore, the presence of homozygote LoF variants in an individual should either be explained by a relaxation of the purifying selection [
52] or by the presence of sequencing errors. In fact, given the evolutionary constraints of LoF, it could be expected that the power for identifying NGS sequencing errors should be enhanced at LoF variants. Sequencing errors typically occur in 0.1–1% of the sequenced nucleotides [
53]; as they are rare events, it can be expected that they will tend to produce rare mutations. Several factors shape the probability of a nucleotide to be erroneously sequenced; in particular, poor-quality bases due to low deep sequencing can be misinterpreted by the sequencers [
17]. It has been shown that even in the whole exome sequencing of the 1000G, there exists variation in depth of sequencing in the different sequencing centers [
54]. Furthermore, there is heterogeneity in the error rates across sequencing platforms [
55]. Given that the different populations were non-homogeneously sequenced at the different centers (
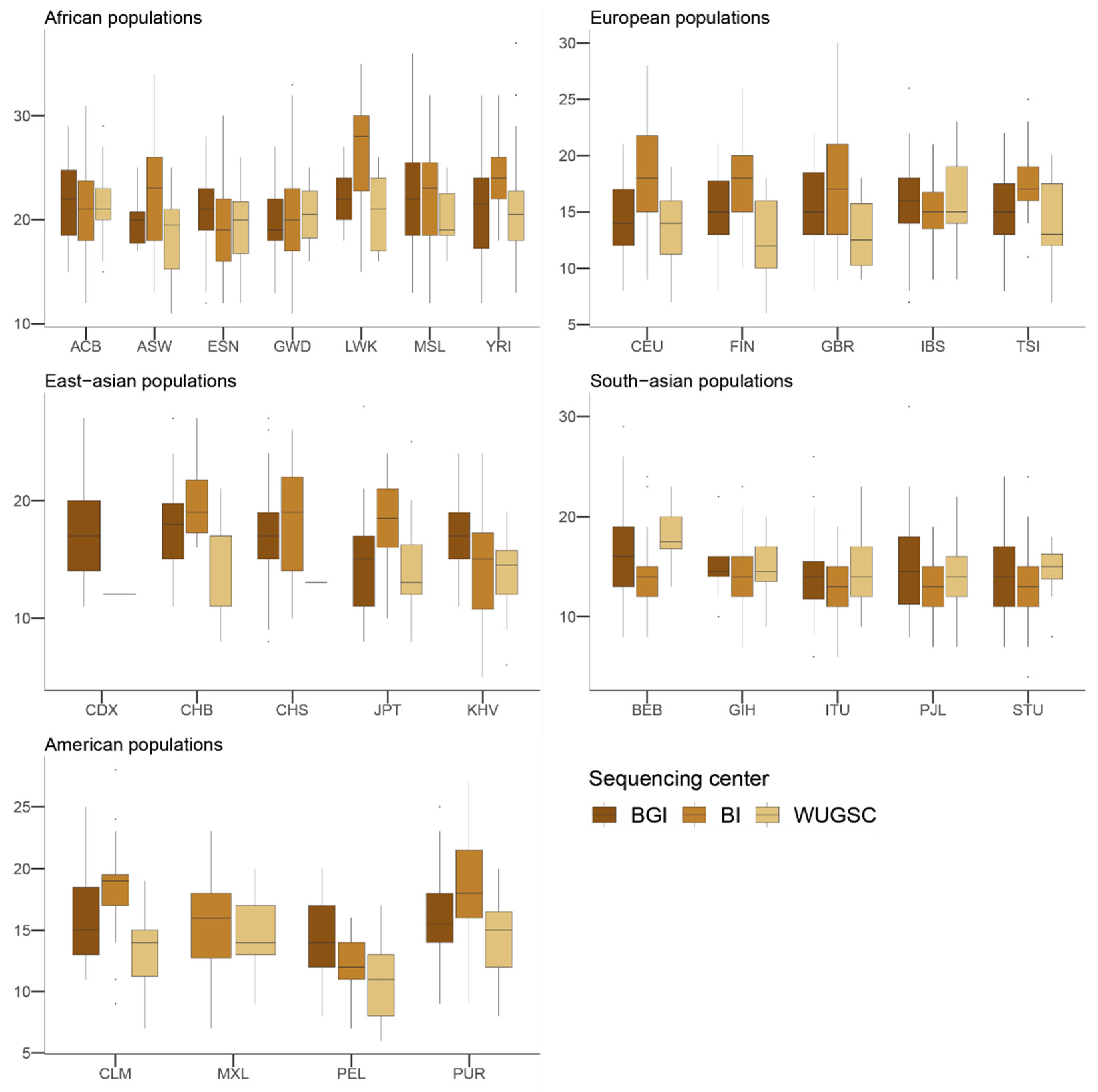
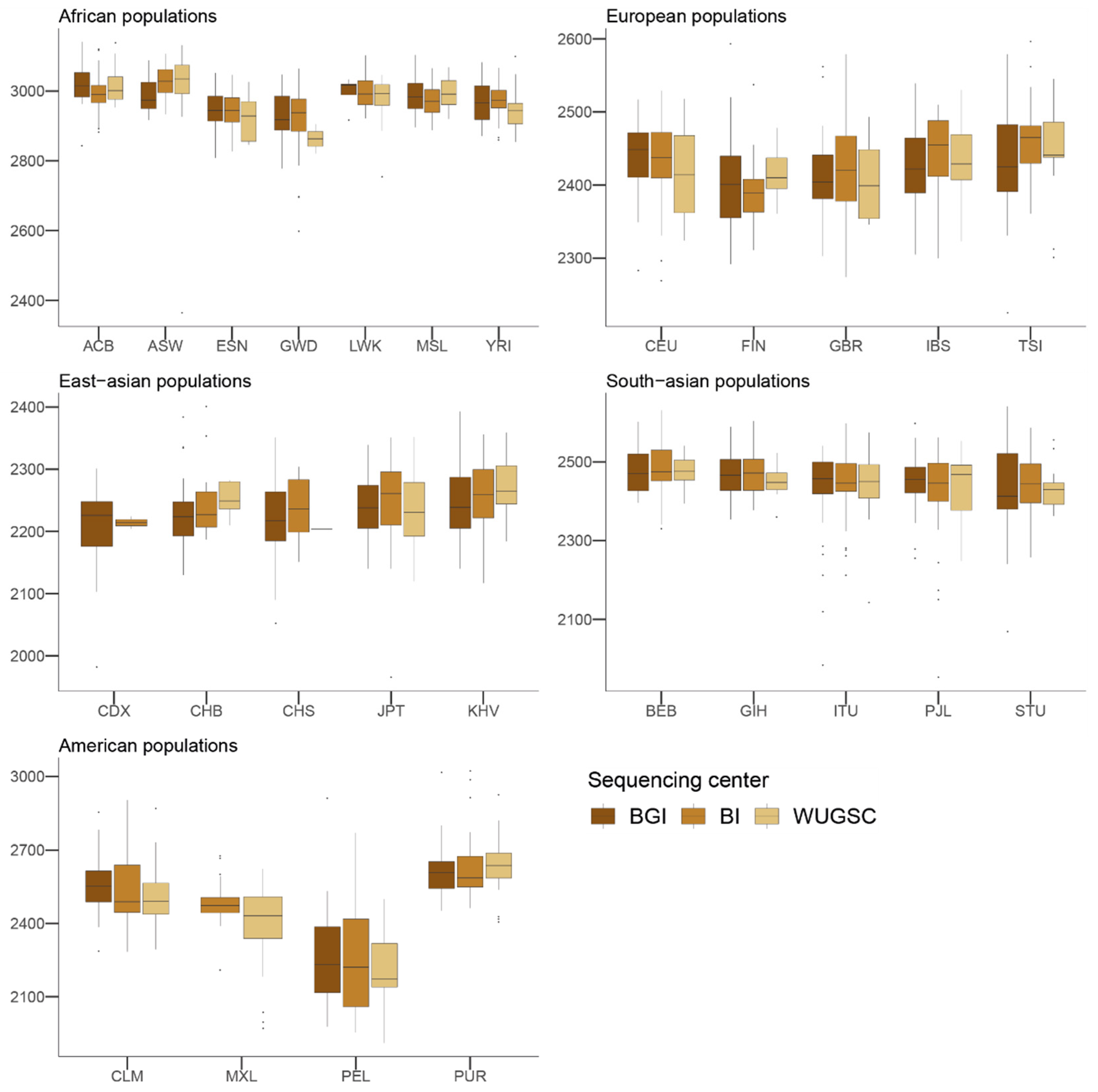
Figure 1) and that each center used a different technologies or methodologies, differences in the LoF fraction within populations and among genetically related populations due to the sequencing center can be expected. In fact, we have observed that for some populations, the number of LoF variants in a sample is directly dependent on the sequencing center used for the different samples. These results suggest a substantial bias in LoF identification due to sequencing center, even when using stringent filters for defining LoF.
Considering the use of the 1000G for calibrating isolated populations and for comparing the presence of LoF (i.e., see [
3]), we wondered to which extent the sequencing center could affect the conclusions regarding the condition of an isolated population. We used the SEP population [
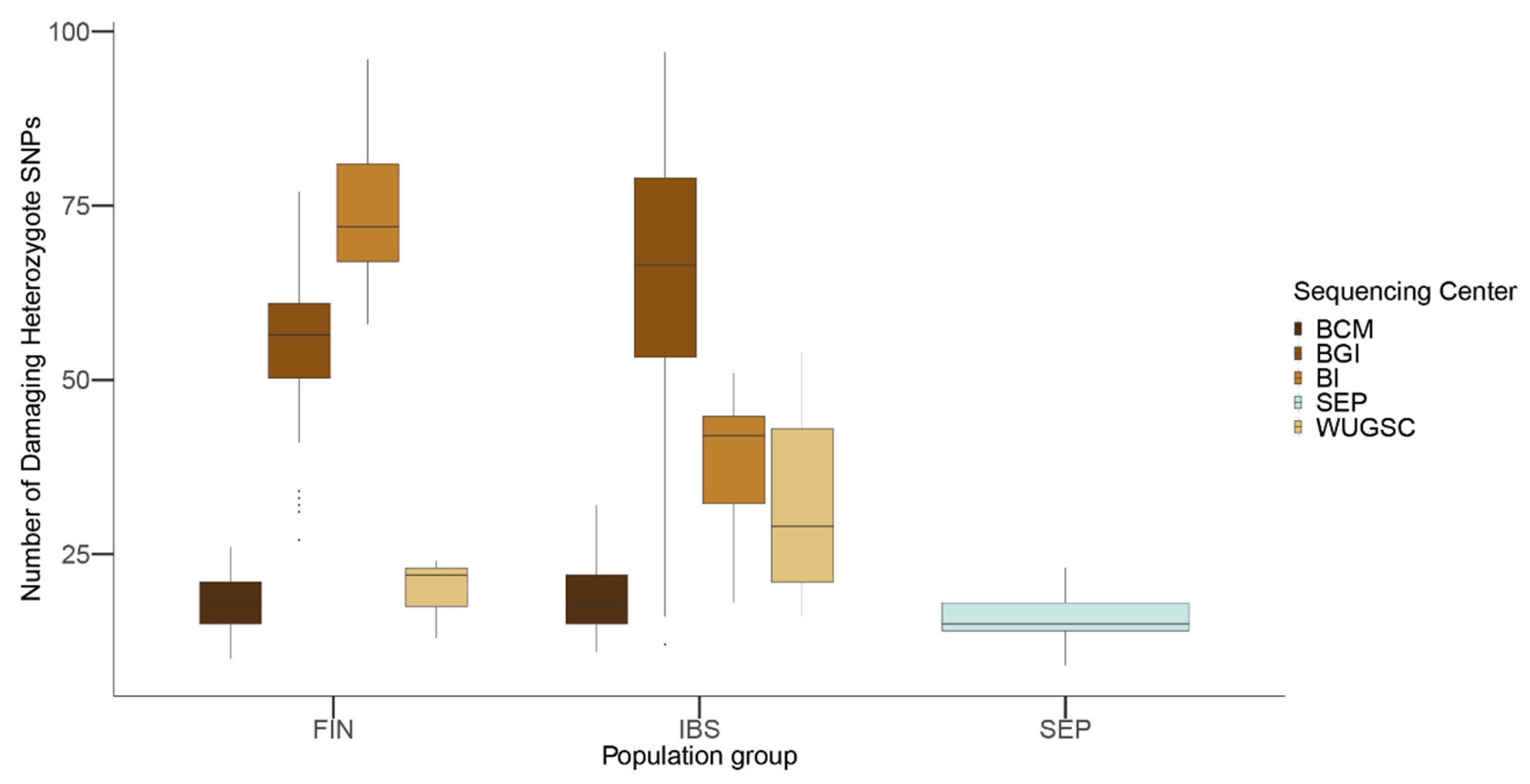
26], which has a similar genetic ancestry to the IBS population and which has been sequenced at ~40× coverage. Compared to IBS and FIN, classically considered isolated populations in Europe showing an excess of private Mendelian diseases [
47], the SEP population has a lower number of LoF damaging alleles compared to both populations. From a biological point of view, this result could be explained by the presence of inbreeding due to isolation and strong purifying selection acting for a long period in the population (as shown with gorilla populations in [
56]). However, when considering the whole genetic variation present in SEP individuals, they genetically resemble IBS individuals and there is no evidence of the presence of long runs of homozygosity or other statistics suggesting long ongoing isolation and inbreeding (see [
26]). Moreover, the observed differences between SEP, FIN, and IBS depend on which sequencing center produced the sequences (
Table 6 and
Table 7). Whereas for BCM the differences in LoF among populations are negligible, for BGI sequenced samples IBS would have an enrichment of LoF variants compared to FIN, which would invalidate previous findings about the population genetics of Finish people [
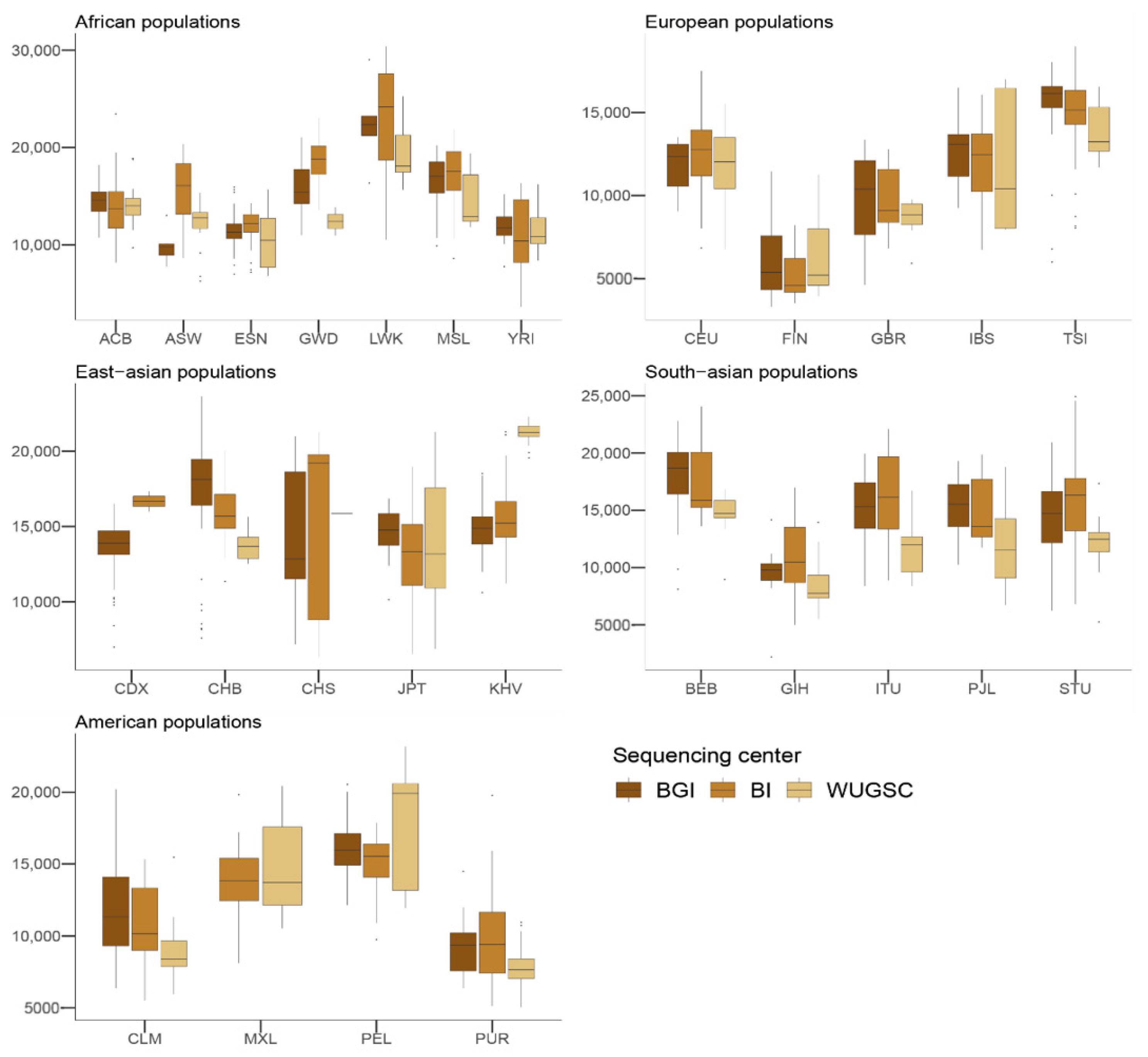
47]. Overall, these results suggest that the observed differences in LoF are more likely due to biases due to the sequencing center. The fact that we observed similar results in the 1000G as those for LoF variants when considering benign mutations suggests that the sequencing bias due to sequencing center could be extended to other mutations occurring at low frequency in these samples. In agreement with this hypothesis, we observed that, for singleton mutations, the sequencing center plays a role, although this signal is not as strong as in the case of LoF variants.
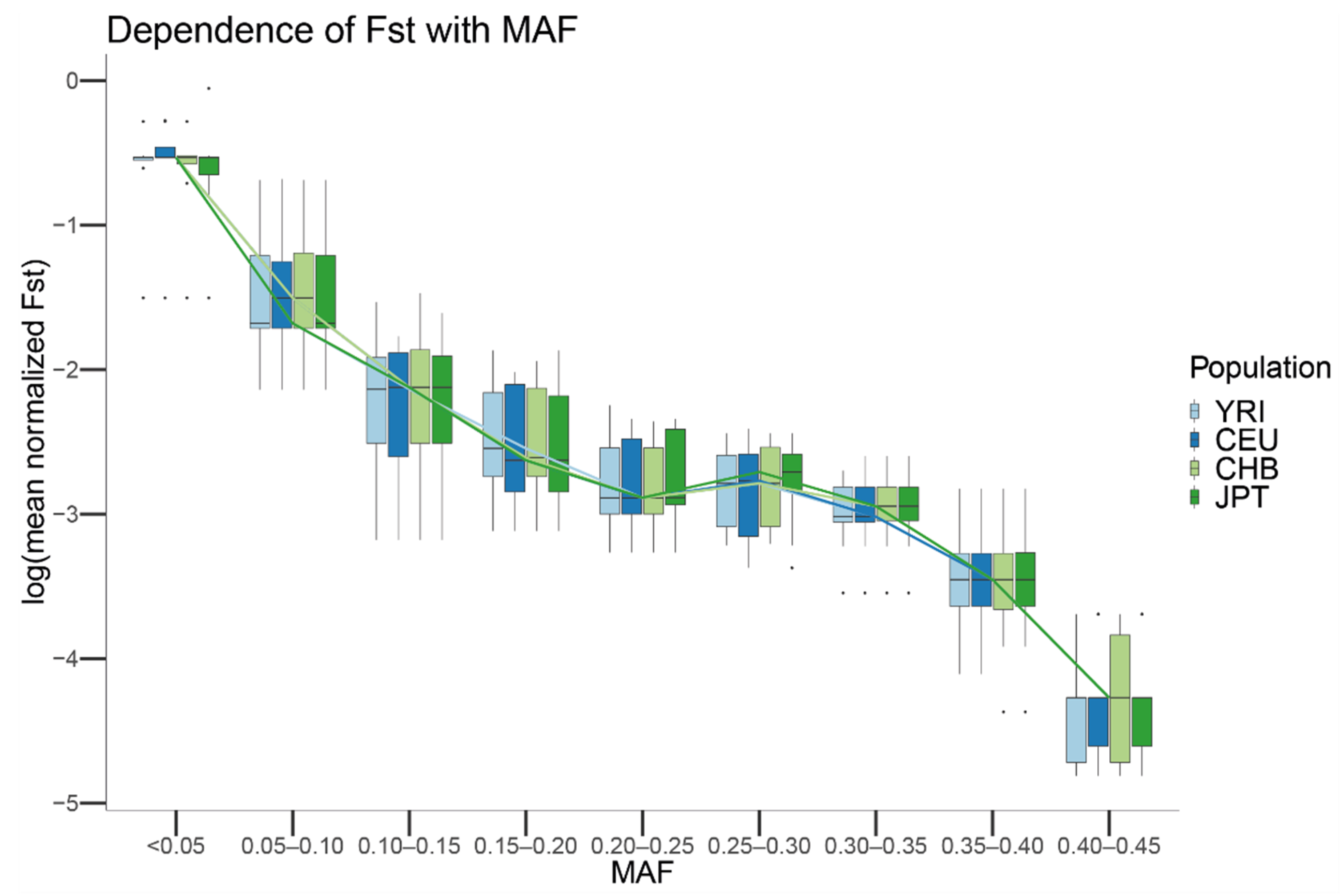
We wondered the limit to which biases in the sequencing center could affect the profile of mutations in a population. Using the originally sampled YRI, CEU, CHB, and JPT populations in Phase 1 of the 1000G, we observed that the bias among centers decreases for all the populations as the frequency of the MAF increases in the considered population. This result could be explained by the variance from a given MAF becoming much higher than the variance due to specific sequencing center error rates, effectively masking their effect. To test this hypothesis, we generated the null distribution of scaled Fsts given a MAF under the hypothesis of no differential sequencing center artifacts. We observed that the Monte Carlo Fst showed the same trend as that observed in the real data.
Overall, these results suggest that the effect we observed in singletons is also observed at higher frequencies. However, when analyzing the detected effect of the sequencing errors on a biological variable that depends on the genetic variation to be detected, such as the level of introgression, we found no effect of the sequencing center on the number of introgressed alleles as defined by [
4]. However, it is interesting to note that strong discrepancies were observed in the amount of introgressed alleles for some populations (CHB and CHS; see
Figure 6). This is particularly relevant because several studies pointed to the presence of heterogeneous patterns of ghost archaic populations in these populations [
4,
25].
5. Conclusions
In this study, we have shown that statistics that use SNVs that occur at low frequencies in the 1000G are influenced by the sequencing center that generated the samples. Since the proportion of samples generated at each center per population is not the same, conclusions about the biological processes that generated these differences can be jeopardized.
The enrichment of putative biases due to batch effects occur mostly with low frequency variants; in particular, in variants where the sequencing error occurs within a gene, it is more likely that the error will generate a ghost functional error in the protein. Nevertheless, the presence of sequencing errors extends to other MAF categories.
Therefore, when considering such particular kind of statistics and genetic variants, caution must be taken in interpreting the results, particularly when merging with another dataset that may have its own batch effects.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}