
First Glimpse into the Genomic Characterization of People from the Imperial Roman Community of Casal Bertone (Rome, First–Third Centuries AD)

 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
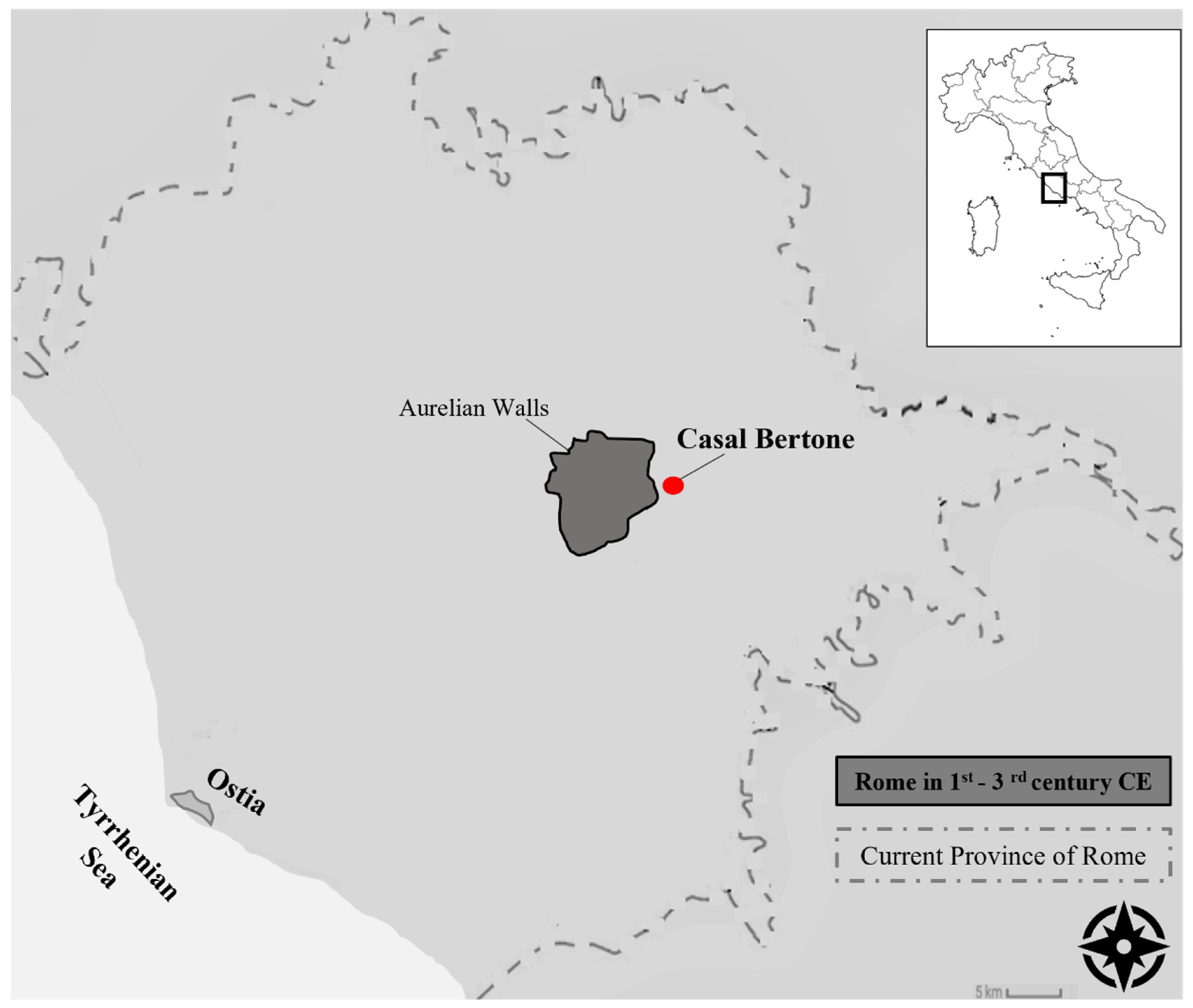
2.1. Samples
2.2. DNA Extraction and Library Preparation
2.3. Data Analysis
3. Results
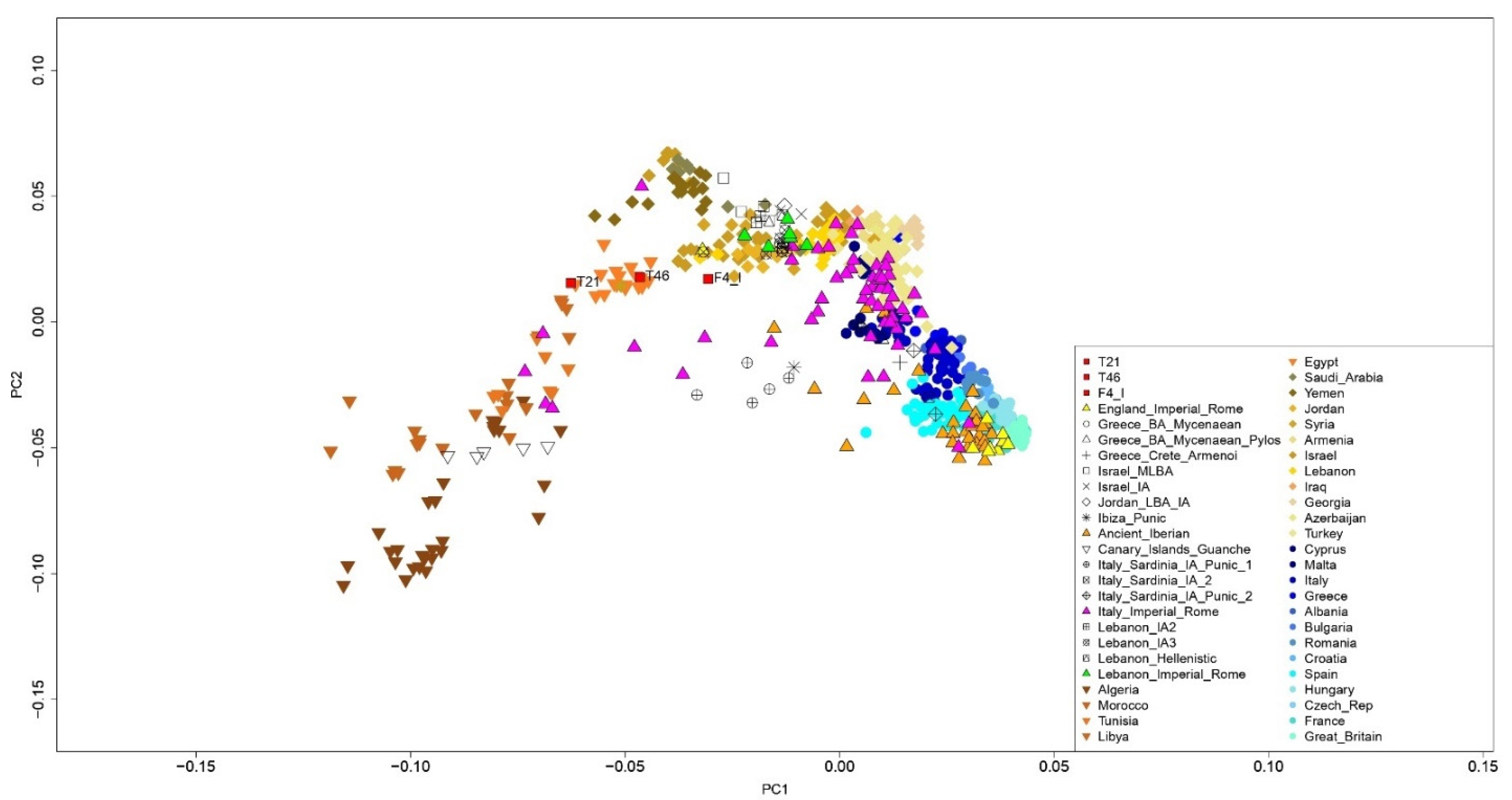
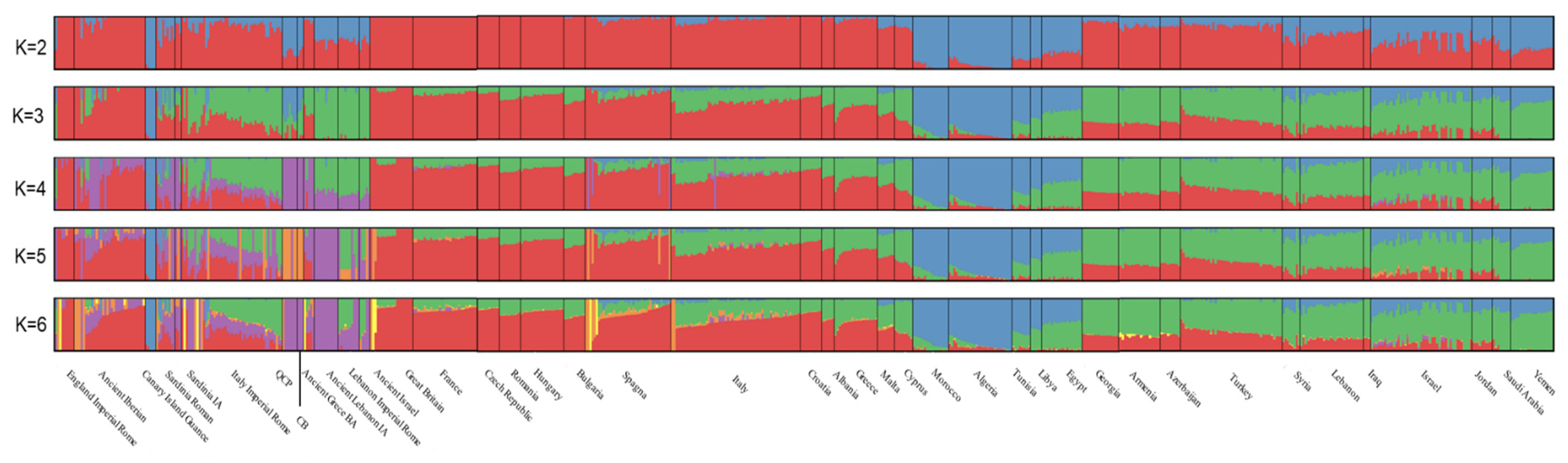
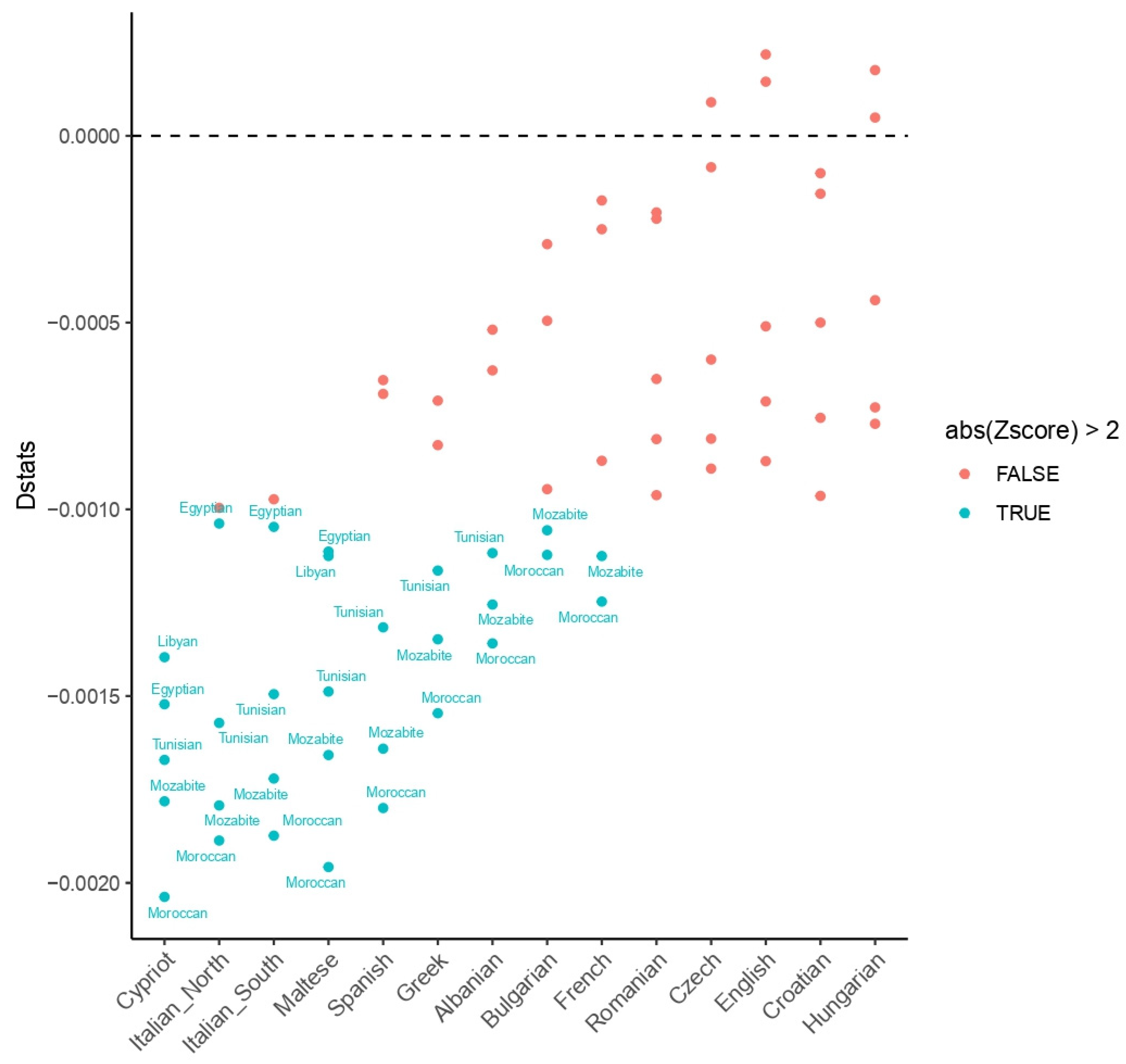
3.1. Human Genetic Profiles
3.2. Microbial Profiles
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vout, C. The Myth of the Toga: Understanding the History of Roman Dress. Greece Rome 1996, 43, 204–220. [Google Scholar] [CrossRef]
- Brøns, C.; Skovmøller, A.; Gisler, J.-R. Colour-Coding The Roman Toga: The Materiality of Textiles Represented in Ancient Sculpture. Antike Kunst 2017, 60, 55–79. [Google Scholar]
- Brain, J.P.; Peabody Museum of Archaeology and Ethnology; Peabody Museum of Salem; Texas Historical Commission. Tunica Treasure; Peabody Museum of Archaeology and Ethnology, Harvard University: Cambridge, MA, USA; Peabody Museum of Salem: Salem, MA, USA, 1979. [Google Scholar]
- Laird, M.L. Review of The World of the Fullo: Work, Economy, and Society in Roman Italy. Phoenix 2014, 68, 378–380. [Google Scholar] [CrossRef]
- Karivieri, A. Ostia as Centre of Production. In Life and Death in a Multicultural Harbour City: Ostia Antica from the Republic through Late Antiquity; Arja Karivieri, Ed.; Institutum Romanum Finlandiae: Rome, Italy, 2020; pp. 209–218. [Google Scholar]
- Bradley, M. It All Comes out in the Was: Looking Harder at the Roman Fullonica. J. Rom. Archaeol. 2002, 15, 20–44. [Google Scholar] [CrossRef]
- Flohr, M. Fullones and Roman society: A reconsideration. J. Rom. Archaeol. 2003, 16, 447–450. [Google Scholar] [CrossRef]
- Ciambelli, S. Le Fulloniche di Pompei. 2017. Available online: https://doi.org/10.13140/RG.2.2.34210.32966 (accessed on 25 September 2021).
- Antonio, M.L.; Gao, Z.; Moots, H.M.; Lucci, M.; Candilio, F.; Sawyer, S.; Oberreiter, V.; Calderon, D.; Devitofranceschi, K.; Aikens, R.C.; et al. Ancient Rome: A genetic crossroads of Europe and the Mediterranean. Science 2019, 366, 708–714. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, F.; Veltre, V.; Romboni, M.; Di Corcia, T.; Scano, G.; Martínez-Labarga, C.; Catalano, P.; Rickards, O. Ancient genomes from a rural site in Imperial Rome (1st–3rd cent. CE): A genetic junction in the Roman Empire. Ann. Hum. Biol. 2021, 48, 234–246. [Google Scholar] [CrossRef]
- Musco, S.; Catalano, P. Tombes d’enfants de l’époque Impériale Dans La Banlieue de Rome: Les Cas de Quarto Cappello Del Prete, de Casal Bertone et de La Nécropole Collatina. In L’enfant et la Mort dans L’antiquité; de Boccard: Paris, France, 2010; pp. 387–402. [Google Scholar]
- Musco, S.; Catalano, P.; Caspio, A.; Pantano, W.; Killgrove, K.; Formoso, E. Le Complexe Archéologique de Casal Bertone. Dossiers d’Archéologie, November/December. 2008; 32–39. [Google Scholar]
- Catalano, P. What Skeletons Tell Us. Med. Secoli 2015, 27, 773–785. [Google Scholar]
- De Angelis, F.; Varano, S.; Battistini, A.; Di Giannantonio, S.; Ricci, P.; Lubritto, C.; Facchin, G.; Brancazi, L.; Santangeli-Valenzani, R.; Catalano, P.; et al. Food for the Empire: Dietary Pattern of Imperial Rome Inhabitants. Evol. Biol. 2020. preprint. [Google Scholar] [CrossRef]
- Caldarini, C.; Zavaroni, F.; Benassi, V. Musculoskeletal Markers, Arthropaty, Traumas. Med. Secoli 2015, 27, 905–967. [Google Scholar]
- Piccioli, A.; Gazzaniga, V.; Catalano, P. Bones: Orthopaedic Pathologies in Roman Imperial Age; Springer: New York, NY, USA, 2016. [Google Scholar]
- Catalano, P.; Giannantonio, S.D.; Pantano, W. Organizzazione Funeraria e Struttura Sociale Degli Inumati Di Casal Bertone (Roma, I–III Sec. d.C.): Ipotesi Antropologiche. In Il Diritto Alla Sepoltura nel Mediterraneo Antico; Bérard, R.-M., Ed.; Collection de l’École française de Rome; Publications de l’École française de Rome: Rome, Italy, 2021; pp. 80–95. [Google Scholar]
- De Angelis, F.; Pantano, W.; Battistini, A. Paleodemographic Analysis. Med. Secoli 2015, 27, 805–872. [Google Scholar]
- Killgrove, K.; Tykot, R.H. Food for Rome: A stable isotope investigation of diet in the Imperial period (1st–3rd centuries AD). J. Anthr. Archaeol. 2013, 32, 28–38. [Google Scholar] [CrossRef]
- Minozzi, S.; Fornaciari, G. Paleopathology of Human Remains. Med. Secoli 2015, 27, 1043–1065. [Google Scholar] [PubMed]
- Mosticone, R.; Pescucci, L.; Porreca, F. Life Conditions: Non-Specific Stress Indicators Additionally, Dentoalveolar Pathologies. Med. Secoli 2015, 27, 969–1042. [Google Scholar] [PubMed]
- Killgrove, K.; Montgomery, J. All Roads Lead to Rome: Exploring Human Migration to the Eternal City through Biochemistry of Skeletons from Two Imperial-Era Cemeteries (1st–3rd c AD). PLoS ONE 2016, 11, e0147585. [Google Scholar] [CrossRef] [PubMed]
- Minozzi, S.; Lunardini, A.; Caldarini, C.; Caramella, D.; Fornaciari, G.; Catalano, P.; Giuffra, V. Metastatic Prostate Carcinoma from Imperial Rome (1st to 2nd Centuries AD). Pathobiology 2018, 85, 289–299. [Google Scholar] [CrossRef]
- Adler, C.J.; Haak, W.; Donlon, D.; Cooper, A. Survival and recovery of DNA from ancient teeth and bones. J. Archaeol. Sci. 2011, 38, 956–964. [Google Scholar] [CrossRef]
- Yang, D.Y.; Eng, B.; Waye, J.S.; Dudar, J.C.; Saunders, S.R. Technical Note: Improved DNA Extraction from Ancient Bones Using Silica-Based Spin Columns. Am. J. Phys. Anthropol. 1998, 105, 539–543. [Google Scholar] [CrossRef]
- Meyer, M.; Kircher, M. Illumina Sequencing Library Preparation for Highly Multiplexed Target Capture and Sequencing. Cold Spring Harb. Protoc. 2010, 2010, pdb-prot5448. [Google Scholar] [CrossRef]
- Li, H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics 2014, 30, 2843–2851. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]
- Skoglund, P.; Northoff, B.H.; Shunkov, M.V.; Derevianko, A.P.; Pääbo, S.; Krause, J.; Jakobsson, M. Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proc. Natl. Acad. Sci. USA 2014, 111, 2229–2234. [Google Scholar] [CrossRef] [PubMed]
- Korneliussen, T.S.; Albrechtsen, A.; Nielsen, R. ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinform. 2014, 15, 356. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Lazaridis, I.; Nadel, D.; Rollefson, G.; Merrett, D.C.; Rohland, N.; Mallick, S.; Fernandes, D.; Novak, M.; Gamarra, B.; Sirak, K.; et al. Genomic insights into the origin of farming in the ancient Near East. Nature 2016, 536, 419–424. [Google Scholar] [CrossRef]
- Günther, T.; Nettelblad, C. The presence and impact of reference bias on population genomic studies of prehistoric human populations. PLoS Genet. 2019, 15, e1008302. [Google Scholar] [CrossRef]
- Patterson, N.; Price, A.L.; Reich, D. Population Structure and Eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef]
- Alexander, D.H.; Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform. 2011, 12, 246. [Google Scholar] [CrossRef]
- Behr, A.A.; Liu, K.Z.; Liu-Fang, G.; Nakka, P.; Ramachandran, S. pong: Fast analysis and visualization of latent clusters in population genetic data. Bioinformatics 2016, 32, 2817–2823. [Google Scholar] [CrossRef]
- Liu, C.-C.; Shringarpure, S.; Lange, K.; Novembre, J. Exploring Population Structure with Admixture Models and Principal Component Analysis. Methods Mol. Biol. 2020, 2090, 67–86. [Google Scholar] [CrossRef]
- Patterson, N.; Moorjani, P.; Luo, Y.; Mallick, S.; Rohland, N.; Zhan, Y.; Genschoreck, T.; Webster, T.; Reich, D. Ancient Admixture in Human History. Genetics 2012, 192, 1065–1093. [Google Scholar] [CrossRef]
- Skoglund, P.; Storå, J.; Gotherstrom, A.; Jakobsson, M. Accurate sex identification of ancient human remains using DNA shotgun sequencing. J. Archaeol. Sci. 2013, 40, 4477–4482. [Google Scholar] [CrossRef]
- Schubert, M.; Ermini, L.; Der Sarkissian, C.; Jónsson, H.; Ginolhac, A.; Schaefer, R.; Martin, M.; Fernández, R.; Kircher, M.; McCue, M.; et al. Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX. Nat. Protoc. 2014, 9, 1056–1082. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Steinberg, K.M.; Larson, D.; Wilson, R.K.; Mardis, E.R. The Next-Generation Sequencing Revolution and Its Impact on Genomics. Cell 2013, 155, 27–38. [Google Scholar] [CrossRef]
- Weissensteiner, H.; Pacher, D.; Kloss-Brandstätter, A.; Forer, L.; Specht, G.; Bandelt, H.-J.; Kronenberg, F.; Salas, A.; Schönherr, S. HaploGrep 2: Mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic Acids Res. 2016, 44, W58–W63. [Google Scholar] [CrossRef] [PubMed]
- Poznik, G.D. Identifying Y-Chromosome Haplogroups in Arbitrarily Large Samples of Sequenced or Genotyped Men. 2016. [Google Scholar]
- Kuhn, J.M.M.; Jakobsson, M.; Günther, T. Estimating genetic kin relationships in prehistoric populations. PLoS ONE 2018, 13, e0195491. [Google Scholar] [CrossRef]
- Ferravante, C.; Memoli, D.; Palumbo, D.; Ciaramella, P.; Di Loria, A.; D’Agostino, Y.; Nassa, G.; Rizzo, F.; Tarallo, R.; Weisz, A.; et al. HOME-BIO (sHOtgun MEtagenomic analysis of BIOlogical entities): A specific and comprehensive pipeline for metagenomic shotgun sequencing data analysis. BMC Bioinform. 2021, 22, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed]
- Elsner, J.; Schibler, J.; Hofreiter, M.; Schlumbaum, A. Burial condition is the most important factor for mtDNA PCR amplification success in Palaeolithic equid remains from the Alpine foreland. Archaeol. Anthr. Sci. 2014, 7, 505–515. [Google Scholar] [CrossRef]
- Mughal, M.R.; DeGiorgio, M. Properties and unbiased estimation of F- and D-statistics in samples containing related and inbred individuals. Genetics 2022, 220, iyab090. [Google Scholar] [CrossRef]
- Tena, D.; Fernández, C.; Lago, M.R. Alcaligenes faecalis: An Unusual Cause of Skin and Soft Tissue Infection. Jpn. J. Infect. Dis. 2015, 68, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Le Maréchal, C.; Fourour, S.; Ballan, V.; Rouxel, S.; Souillard, R.; Chemaly, M. Detection of Clostridium botulinum group III in environmental samples from farms by real-time PCR using four commercial DNA extraction kits. BMC Res. Notes 2018, 11, 441. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.G.; Hurtado, R.; Gomes, L.G.R.; Profeta, R.; Rifici, C.; Attili, A.R.; Spier, S.J.; Mazzullo, G.; Morais-Rodrigues, F.; Gomide, A.C.P.; et al. Complete genome analysis of Glutamicibacter creatinolyticus from mare abscess and comparative genomics provide insight of diversity and adaptation for Glutamicibacter. Gene 2020, 741, 144566. [Google Scholar] [CrossRef] [PubMed]
- Baraboutis, I.G.; Tsagalou, E.P.; Lepinski, J.L.; Papakonstantinou, I.; Papastamopoulos, V.; Skoutelis, A.T.; Johnson, S. Primary Staphylococcus aureus urinary tract infection: The role of undetected hematogenous seeding of the urinary tract. Eur. J. Clin. Microbiol. Infect. Dis. 2010, 29, 1095–1101. [Google Scholar] [CrossRef][Green Version]
- Flohr, M. Fulling. Available online: https://oxfordre.com/classics/view/10.1093/acrefore/9780199381135.001.0001/acrefore-9780199381135-e-8183 (accessed on 1 November 2021). [CrossRef]
- Sigismund-Nielsen, H. Slave and Lower-Class Roman Children. Available online: https://www.oxfordhandbooks.com/view/10.1093/oxfordhb/9780199781546.001.0001/oxfordhb-9780199781546-e-014 (accessed on 22 November 2021). [CrossRef]
- Shahandeh, Z.; Shafi, H.; Sadighian, F. Association of staphylococcus cohnii subspecies urealyticum infection with recurrence of renal staghorn stone. Caspian J. Intern. Med. 2015, 6, 40–42. [Google Scholar] [PubMed]
- Lozano, V.; Fernández, G.; Spencer, P.L.; Taylor, S.L.; Hatch, R. Staphylococcus epidermidis in Urine Is Not Always Benign: A Case Report of Pyelonephritis in a Child. J. Am. Board Fam. Med. 2015, 28, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Orrett, F.A.; Shurland, S.M. Significance of coagulase-negative staphylococci in urinary tract infections in a developing country. Conn. Med. 1998, 62, 199–203. [Google Scholar]
- Pati, A.; LaButti, K.; Pukall, R.; Nolan, M.; Del Rio, T.G.; Tice, H.; Cheng, J.-F.; Lucas, S.; Chen, F.; Copeland, A.; et al. Complete genome sequence of Sphaerobacter thermophilus type strain (S 6022T). Stand. Genom. Sci. 2010, 2, 49–56. [Google Scholar] [CrossRef]
- Wang, X.; Xue, Y.; Yuan, S.; Zhou, C.; Ma, Y. Salinicoccus halodurans sp. nov., a moderate halophile from saline soil in China. Int. J. Syst. Evol. Microbiol. 2008, 58, 1537–1541. [Google Scholar] [CrossRef][Green Version]
- Shivaji, S.; Suresh, K.; Chaturvedi, P.; Dube, S.; Sengupta, S. Bacillus arsenicus sp. nov., an arsenic-resistant bacterium isolated from a siderite concretion in West Bengal, India. Int. J. Syst. Evol. Microbiol. 2005, 55 Pt 3, 1123–1127. [Google Scholar] [CrossRef]
- McLaughlin, R. The Roman Empire and the Indian Ocean: The Ancient World Economy and the Kingdoms of Africa, Arabia and India; Pen & Sword Military: Barnsley, UK, 2014. [Google Scholar]
- Essahale, A.; Malki, M.; Marín, I.; Moumni, M. Bacterial diversity in Fez tanneries and Morocco’s Binlamdoune River, using 16S RNA gene based fingerprinting. J. Environ. Sci. 2010, 22, 1944–1953. [Google Scholar] [CrossRef]
- Chowdhary, P.; More, N.; Raj, A.; Bharagava, R.N. Characterization and identification of bacterial pathogens from treated tannery wastewater. Microbiol. Res. Int. 2017, 5, 30–36. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Age | Area | Skeletal District |
|---|---|---|---|
| T9 | 18 months ± 3 | Necropolis | Petrous bone |
| T21 | 6/9 months | Area Q | Petrous bone and femur |
| T46 | 2 years ± 8 months | Area Q | Petrous bone |
| F4_I | 4/9 years | Mausoleum | Petrous bone and femur |
| F9 | 9/15 years | Mausoleum | Femur and tooth |
| Sample Name | Number of Reads | Coverage (Mean) | Coverage (SD) | Non-Missing Calls vs. 1240 K Panel |
|---|---|---|---|---|
| T21 | 2,342,021 | 0.0562 | 0.2982 | 31,972 |
| F4_I | 2,029,640 | 0.0458 | 0.2662 | 27,178 |
| T46 | 1,267,711 | 0.0311 | 0.2228 | 17,522 |
| Reads to This Taxon | NCBI ID | Indented Scientific Name |
|---|---|---|
| T21 | ||
| 12,050 | 511 | Alcaligenes faecalis |
| 6096 | 507 | Alcaligenes |
| 2482 | 255247 | Fictibacillus arsenicus |
| 2031 | 110319 | Nocardioides sp. CF8 |
| 1803 | 929704 | Myroides odoratus DSM 2801 |
| 1182 | 1485 | Clostridium |
| 969 | 1329200 | Fictibacillus |
| 886 | 286 | Pseudomonas |
| 878 | 1386 | Bacillus |
| 715 | 1471761 | Novibacillus thermophilus |
| 696 | 292459 | Symbiobacterium thermophilum IAM 14863 |
| 642 | 1123519 | Pseudomonas stutzeri DSM 10701 |
| 637 | 269800 | Thermobifida fusca YX |
| 560 | 717605 | Thermobacillus composti KWC4 |
| 559 | 1491 | Clostridium botulinum |
| T46 | ||
| 13,362 | 1873 | Micromonospora |
| 10,458 | 286 | Pseudomonas |
| 9684 | 1123519 | Pseudomonas stutzeri DSM 10701 |
| 5943 | 129337 | Geobacillus |
| 4711 | 255247 | Fictibacillus arsenicus |
| 3250 | 1883 | Streptomyces |
| 1862 | 613 | Serratia |
| 1804 | 1329200 | Fictibacillus |
| 1349 | 1007105 | Pusillimonas sp. T7-7 |
| 1259 | 1471761 | Novibacillus thermophilus |
| 1244 | 1491 | Clostridium botulinum |
| 1078 | 316 | Pseudomonas stutzeri |
| 990 | 1747 | Cutibacterium acnes |
| 976 | 511 | Alcaligenes faecalis |
| 883 | 1196031 | Bacillus oceanisediminis 2691 |
| F4_I | ||
| 750 | 286 | Pseudomonas |
| 455 | 1279 | Staphylococcus |
| 385 | 1280 | Staphylococcus aureus |
| 310 | 469 | Acinetobacter |
| 291 | 382 | Sinorhizobium meliloti |
| 251 | 479434 | Sphaerobacter thermophilus DSM 20745 |
| 181 | 305 | Ralstonia solanacearum |
| 174 | 407035 | Salinicoccus halodurans |
| 173 | 1485 | Clostridium |
| 170 | 358 | Agrobacterium tumefaciens |
| 165 | 861360 | Glutamicibacter arilaitensis Re117 |
| 144 | 1386 | Bacillus |
| 114 | 357 | Agrobacterium |
| 114 | 1712675 | Turicibacter sp. H121 |
| 111 | 1301 | Streptococcus |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Angelis, F.; Romboni, M.; Veltre, V.; Catalano, P.; Martínez-Labarga, C.; Gazzaniga, V.; Rickards, O. First Glimpse into the Genomic Characterization of People from the Imperial Roman Community of Casal Bertone (Rome, First–Third Centuries AD). Genes 2022, 13, 136. https://doi.org/10.3390/genes13010136
De Angelis F, Romboni M, Veltre V, Catalano P, Martínez-Labarga C, Gazzaniga V, Rickards O. First Glimpse into the Genomic Characterization of People from the Imperial Roman Community of Casal Bertone (Rome, First–Third Centuries AD). Genes. 2022; 13(1):136. https://doi.org/10.3390/genes13010136
Chicago/Turabian StyleDe Angelis, Flavio, Marco Romboni, Virginia Veltre, Paola Catalano, Cristina Martínez-Labarga, Valentina Gazzaniga, and Olga Rickards. 2022. "First Glimpse into the Genomic Characterization of People from the Imperial Roman Community of Casal Bertone (Rome, First–Third Centuries AD)" Genes 13, no. 1: 136. https://doi.org/10.3390/genes13010136
APA StyleDe Angelis, F., Romboni, M., Veltre, V., Catalano, P., Martínez-Labarga, C., Gazzaniga, V., & Rickards, O. (2022). First Glimpse into the Genomic Characterization of People from the Imperial Roman Community of Casal Bertone (Rome, First–Third Centuries AD). Genes, 13(1), 136. https://doi.org/10.3390/genes13010136