Abstract
Adverse exposures during pregnancy have been shown to contribute to susceptibility for chronic diseases in offspring. Maternal diabetes during pregnancy is associated with higher risk of pregnancy complications, structural birth defects, and cardiometabolic health impairments later in life. We showed previously in a mouse model that the placenta is smaller in diabetic pregnancies, with reduced size of the junctional zone and labyrinth. In addition, cell migration is impaired, resulting in ectopic accumulation of spongiotrophoblasts within the labyrinth. The present study had the goal to identify the mechanisms underlying the growth defects and trophoblast migration abnormalities. Based upon gene expression assays of 47 candidate genes, we were able to attribute the reduced growth of diabetic placenta to alterations in the Insulin growth factor and Serotonin signaling pathways, and provide evidence for Prostaglandin signaling deficiencies as the possible cause for abnormal trophoblast migration. Furthermore, our results reinforce the notion that the exposure to maternal diabetes has particularly pronounced effects on gene expression at midgestation time points. An implication of these findings is that mechanisms underlying developmental programming act early in pregnancy, during placenta morphogenesis, and before the conceptus switches from histiotrophic to hemotrophic nutrition.
1. Introduction
Diabetes during pregnancy is a known risk factor for congenital defects and pregnancy complications, such as spontaneous abortions and preeclampsia [1,2,3,4]. Progeny from diabetic pregnancies also have a higher risk for metabolic disease later in life [5,6,7], which may be mediated by lower or higher than average weight at birth, by fetal hyperinsulinemia [8], or by as yet unidentified mechanisms of developmental programming [9,10].
In a mouse model of type I diabetes, we previously showed that fetal growth is reduced, noticeable after gestational day 15.5, and that the growth reduction was more pronounced when the dam was fed a diet with higher fat and lower protein content compared to chow [11]. While placenta size was not altered by chow diet, placental growth was impaired during diabetic pregnancy with the higher fat content diet [12]. In particular, the spongiotrophoblast layer and the labyrinth area were significantly reduced in thickness by gestational days 15.5 and 18.5, respectively [13]. The present study was therefore designed to investigate—in females fed the higher fat content diet—the effects of diet on the expression of a number of genes implicated in placenta growth and fetal growth, including Insulin-like growth factors (Igfs), and their receptor and binding protein genes.
In addition, even with normal diet, the contribution of spongiotrophoblast cells to the junctional zone of the placenta was decreased in diabetic pregnancies, and cells expressing the spongiotrophoblast marker Trophoblast-specific protein alpha (TpbpA) ectopically accumulated in the labyrinth [13]. This suggested to us that cell differentiation and cell migration is altered in the placenta under conditions of maternal hyperglycemia, and, potentially, affected by maternal diet. As migration and invasion of the maternal decidua by trophoblasts requires remodeling of the extracellular matrix, the present study also examines whether the impaired trophoblast migration in diabetic placentae could be mediated by altered expression of metalloproteases or their inhibitors. Furthermore, we wanted to assess to what extent maternal diet affects the expression of these genes in the placenta. A crucial link between metalloproteinase expression and Insulin-like growth factor signaling are the pregnancy-associated plasma proteins metalloproteases which are involved in the release of bioactive Igf from Igf-binding proteins.
Because metalloproteinase action can be a sign of inflammation, we also investigated the expression of genes involved in altered immune responses and inflammation, including genes in the prostaglandin pathway. Prostaglandins are well-known mediators of inflammation, and play a critical role in the timing of implantation and parturition [14]. It is known that prostaglandin signaling affects trophoblast invasion [15,16], and, therefore, we reasoned that altered prostaglandin signaling might contribute to the impaired placenta development in diabetic pregnancies. The selection of genes to be investigated in this study was guided by our earlier gene expression profiling results that had demonstrated their expression in normal and diabetic placentae [13].
2. Materials and Methods
Diabetes was induced in FVB mice by injection of Streptozotocin, as described previously [11,12]. The blood glucose levels and morphometric parameters of pregnant dams have been published [11,12]. Archived placenta samples were used in the present study that had been collected and processed for RNA extraction as published, and quantitative PCR was performed exactly as before [11,12,17,18].
Diets consisted of chow diet (Labdiet Purina 5001) until FVB females were randomly assigned at eight weeks of age to either continue feeding on chow, or breeder diet (Purina 5015), which is formulated specifically to support pregnancy and lactation. The compositions of the diets are described in the manufacturer’s datasheets (see Supplemental Files) and differ in protein and fat content, with minor variation in carbohydrate content. Importantly, both diets are nutritionally complete for mice in normal metabolic conditions. Females consumed the respective diet for at least four weeks before mating.
Primers for real-time quantitative PCR amplification were designed following the criteria described in Kruger et al. (2006) [17]. The gene encoding DNA Polymerase epsilon 4 was used as the reference gene for normalization. Where possible, primers were positioned so that the resulting amplicon would span an exon–exon junction so that amplicons would be derived only from cDNA templates. Sequences of primers and their coordinates are listed in Table 1. Primers for genes assayed in the current study but not included on this list have been published previously [12].

Table 1.
PCR primers employed in this study and their amplification efficiencies. Gene symbols follow the Mouse Genome Informatics (http://www.informatics.jax.org accessed on 31 October 2021) nomenclature: Ascl2: Achaete-scute family bHLH transcription factor 2; Bmp2: Bone morphogenetic protein 2; Calcb: Calcitonin-related polypeptide, beta; Cbr1: Carbonyl reductase 1; Cck: Cholecystokinin; Ctsq: Cathepsin Q; Gpx4: Glutathione peroxidase 4; Htr2b: 5-Hydroxytryptamine (serotonin) receptor 2B; Igf1: Insulin-like growth factor 1; Igf1r: Insulin-like growth factor 1 receptor; Igf2: Insulin-like growth factor 2; Igf2r: Insulin-like growth factor 2 receptor; Igfbp3: Insulin-like growth factor binding protein 3; Igfbp5: Insulin-like growth factor binding protein 5; Kazald1: Kazal-type serine peptidase inhibitor domain 1; Klhl5: Kelch-like 5; Klk8: Kallikrein related-peptidase 8; Mmp1a: Matrix metallopeptidase 1a (interstitial collagenase); Mmp13: Matrix metallopeptidase 13; Mmp14: Matrix metallopeptidase 14 (membrane-inserted); Mmp15: Matrix metallopeptidase 15; Mmp2: Matrix metallopeptidase 2: Mmp3: Matrix metallopeptidase 3; Mmp8: Matrix metallopeptidase 8; Mmp9: Matrix metallopeptidase 9; Pappa: Pregnancy-associated plasma protein A; Pappa2: Pappalysin 2; Pla2g4a: Phospholipase A2, group IVA (cytosolic, calcium-dependent); Pla2g5: Phospholipase A2, group V; Ptges: Prostaglandin E synthase; Ptges3: Prostaglandin E synthase 3; Ptgs1: Prostaglandin-endoperoxide synthase 1; Rbbp9: Retinoblastoma binding protein 9, serine hydrolase; Rnase2a: Ribonuclease, RNase A family, 2A (liver, eosinophil-derived neurotoxin); Sfrp5: Secreted frizzled-related sequence protein 5; Slco2a1: Solute carrier organic anion transporter family, member 2a1; Spi16: Serine protease inhibitor 16; Spink8: Serine peptidase inhibitor, Kazal type 8.
Our method for quantitation of gene expression levels was applied exactly as in previous studies [11,12,17,18]. Measurements on each sample were done in triplicates and obtained values were averaged. Amplification efficiencies were calculated, and statistical evaluation was performed on ∆CT values. Experimental groups consisted of six placentae, each from a separate diabetic or control pregnancy, respectively [12,18]. Figures show inverse ∆CT to depict the highest expression as the highest visual points on the graphs.
Two-tailed T-tests—with assumption of unequal variance—were performed to evaluate significance of differences in gene expression levels between groups; a p-value of <0.05 was considered significant.
In situ hybridizations were performed on frozen sections as described [13], with most probes obtained from Open Biosystems/Thermo Fisher.
3. Results and Discussion
Our prior studies provided evidence that in diabetic mouse pregnancies, placentae are smaller and that their junctional zones are thinner, when compared to placentae from metabolically normal pregnancies [13]. We also showed that maternal diabetes affects the migration of spongiotrophoblast cells, some of which form ectopic cell clusters in the labyrinth instead of contributing to the junctional layer (Figure 1).
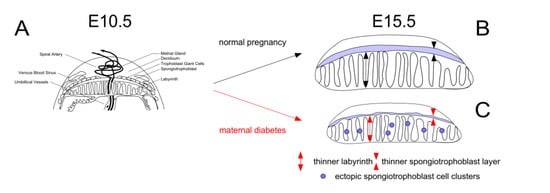
Figure 1.
Overview of placental anomalies in mouse diabetic pregnancies.Panel A: Schematic depiction of the placenta at E10.5. Compared to normal pregnancies (Panel B), the placenta remains smaller in diabetic pregnancies (Panel C), with reduced thickness of labyrinth and junctional zone, and ectopic accumulation of spongiotrophoblasts in the labyrinth, due to impaired cell migration. Placenta weights were lowest in diabetic pregnancies where the dam was fed a diet (Breeder diet) that is optimized to promote pregnancy and lactation under normal conditions.
The reductions in placental size are more pronounced in diabetic pregnancies when dams are fed breeder diet compared to chow [12]. Taken together, these observations prompted us to investigate the potential underlying mechanisms, focusing on genes known to be involved in embryonic and placental growth, trophoblast invasion, cell migration, and inflammatory processes. Because we showed earlier that the growth differences and placental cellular abnormalities in late pregnancy are preceded by alterations in gene expression at midgestation [12,13], we assayed placental mRNA levels at various gestational time points.
Expression of 47 genes was measured in placenta samples from normal and diabetic pregnancies, where dams had been fed either chow or breeder diet for four weeks prior to mating. In all graphs presented here, chow-fed experimental groups are represented by open symbols and dashed lines, while breeder diet-fed groups are depicted by closed symbols and solid lines; the diabetic condition is always marked in red.
The Insulin growth factor signaling system has long been known to regulate embryonic growth and placental size in the mouse [19]. However, as Figure 2 shows, no significant differences were detected in the expression levels of genes encoding Igf 1, Igf2, or their respective receptors, nor the two Igf-binding proteins known to be expressed in mouse placenta. Pregnancy-associated protein A (Pappa) transcript levels were lower in diabetic placentae compared to controls, but only at E12.5; Pappa2 levels were also lower with maternal diabetes, from E9.5 on, with both diets.
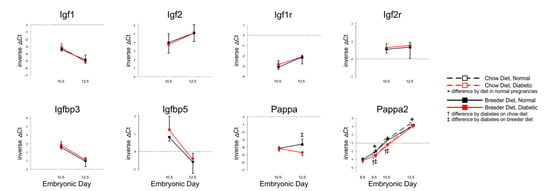
Figure 2.
Expression of Igfs, their receptors, Igf-binding proteins and their proteases. Quantitative PCR measurements were performed at various time points, in dams that were either fed chow or breeder diet, and became diabetic in the week before, or were normoglycemic at the time of mating.
These results suggest that reduced expression of Pappa proteins could play a role in the growth deficiency of placentae under diabetic conditions, as Pappa metalloproteases have Igf-binding proteins as their main targets [20,21], and reduced proteolytic activity would result in reduced Igf availability [22]. Consistent with this proposition is the finding that targeted disruption of the Pappa gene in mice causes intrauterine growth restriction [23]. In the case of Pappa2 deficiency, growth retardation was only evident postnatally [24], but this does not exclude the possibility that Pappa2 reduction in diabetic pregnancies could influence postnatal growth trajectories, as offspring from mouse diabetic pregnancies are born smaller [11], and intrauterine growth-restricted progeny can experience substantial catch-up growth [25,26].
We then investigated several genes that are known to be required for embryonic development from defects reported with targeted disruption, such as for example embryonic lethality in mice with targeted disruption of Proprotein convertase subtilisin/kexin type 5 (Pcsk5) [27] and Bone morphogenetic protein 2 (Bmp2) [28], multiple defects in mutants for endoplasmatic reticulum metallopeptidase Ermp1 [29], Calcitonin related peptide beta (Calcb) [30], Mmp14 [31], and overall growth delay as in mutants for the Tgf-beta induced Tgfbi gene [32]. Atonal bHLH transcription factor 8 (Atoh8/Math6)-deficient mice exhibit embryonic lethality, due to placental defects [33], which are also suspected but as of yet uncharacterized in many embryonic and early postnatal lethal mouse mutants [34]. In addition, we assayed genes with documented expression in specific placental cell types, such as Ascl2 in the labyrinth and spongiotrophoblasts [35], the Slc6a4 serotonin transporter in trophoblast giant cells [36], TpbpA in spongiotrophoblasts [37], Cck in cells surrounding the spiral arteries [13], and the prolactin Prl5a1, which is expressed in spongiotrophoblasts [38].
As shown in Figure 3, many of these genes exhibited significantly different expression levels in placentae from diabetic pregnancies at midgestation, i.e., E10.5, while differences at later stages were detected only for Cck and Slc6a4. Higher expression of Cck was observed in breeder-diet-fed dams, but differences between normal and diabetic placentae were no longer present after E10.5, after which diet appeared to be the main driver. Slc6a4 displayed a similar pattern, at a moderate level of expression up until E12.5, but its expression was significantly lower in diabetic placentae by E15.5. Although transcripts for the Serotonin receptor Htr2b only differed at E10.5, the decreased expression of the Serotonin transporter Slc6a4 suggests that placental serotonin signaling could be impaired at later stages of gestation. Since Serotonin acts as a growth factor during development [39,40,41,42], reduced availability in the second half of pregnancy may contribute to the smaller placenta size in diabetic pregnancies. Furthermore, it was recently proposed that Serotonin may control placental nutrient uptake [43].
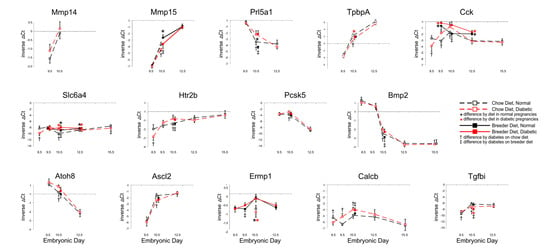
Figure 3.
Expression of genes with roles in embryonic and placental development. Quantitative PCR measurements were performed at various time points, in dams that were either fed chow or breeder diet, and became diabetic in the week before, or were normoglycemic at the time of mating.
However, from some of the temporal trajectories it is not intuitively obvious how changes in expression of these genes could explain the reduced growth of the placenta after E12.5, particularly in diabetic breeder-diet-fed dams. It cannot be excluded that the observed changes at midgestation (E10.5, or even earlier) could subsequently influence signaling pathways that were not included in our investigations. It is important to note here that technical factors are unlikely to account for the large number of genes with differences at E10.5, as both higher and lower transcript levels of individual genes were measured in diabetic placentae compared to normal. Furthermore, gene expression in distinct cell types may respond uniquely to diet and maternal diabetes. Likewise, the timing of responses to metabolic conditions may also be cell-type-specific. As embryonic blood circulation is believed not to become fully established until E10 [44], the transcriptional responses of placental genes at E10.5 could also reflect the newly developed capacity for, and the consequences of, oxidative metabolism in the embryo, after the switch from histotrophic to hemotrophic nutrition. Furthermore, we have previously shown that the responses of placental genes to influences of diet and maternal metabolic disease follow diverse patterns—with reactivity to either one of these conditions or both conditions combined—that produce additive as well as antipodal effects on expression levels [12]. Taken together, these considerations suggest that likely multiple pathways, possibly in different phases of gestation, contribute to reduced placenta size in diabetic pregnancies.
Signaling pathways that are known or could be suspected to be involved in decidualization and in trophoblast invasion of the decidua, or in trophoblast migration were also assayed. Prostaglandin signaling has been implicated in these processes previously [45,46,47,48,49]. Prostanoid signaling molecules are produced from the polyunsaturated fatty acid Arachidonic acid, which is released from phospholipids by phospholipases, such as Lipoprotein lipase (Lpl) and the Pla2 enzymes. Various enzymatic steps can then produce prostanoids with pro- or anti-inflammatory properties. Potential aberrations in these processes in diabetic pregnancies are suggested by prior evidence that prostanoid production is altered in diabetic rat placenta [50], and that supplementation of Arachidonic acid and/or Prostaglandins can reduce the incidence of developmental defects in rodent diabetic pregnancies [51,52,53,54,55]. Hydroxy-prostaglandindehydrogenase (Hpgd) and Carbonylreductase (Cbr1) are two enzymes controlling prostaglandin availability, as does the Slco2a1 prostaglandin transporter [14], and Glutathione peroxidase 4 (Gpx4) is critically required to eliminate lipid radicals that form when unsaturated fatty acids are involved in lipid peroxidation under conditions of oxidative stress [56]. Elevated oxidative stress has been documented in placentae from rodent diabetic pregnancies [50].
Figure 4 displays gene expression measurements for several of the enzymes involved in the Arachidonic acid–Prostaglandin pathway. Intriguingly, Lpl expression was found to be lower in diabetic pregnancies, but there were no differences in Phospholipase 2g4a (Pla2g4) nor Prostaglandin endoperoxide synthase (Ptsg1/Cox-1) expression. While not altered by maternal diabetes, Ptgs2 (Cox-2) levels were elevated when the dam was fed breeder diet, possibly indicative of increased prostanoid production under these dietary conditions. Unaltered Gpx4 expression, at least at the E10.5 time point measured, did not provide evidence this would be associated with increased lipid peroxidation. Phospholipase 2g5 transcript levels were elevated by diet, and by maternal diabetes, too, but only at E10.5. Of the Prostaglandin E synthesizing enzymes, only Ptges2 levels were changed, reduced by diet, and reduced by maternal diabetes. While Cbr1 levels did not change, the apparent increase of Slco2a1 prostaglandin transporter expression in diabetic conditions was not significant, due to large variations between individuals within each experimental group. Intriguingly, Hpgd levels were higher with breeder diet feeding, and in diabetic conditions. This was also the case for expression of Cyp1a1, which can metabolize a wide variety of polyunsaturated lipids and their derivatives, including Arachidonic acid [57]. Taken together, these results would be consistent with reduced synthesis and increased elimination of Arachidonic acid and Prostaglandin E2 in diabetic conditions.
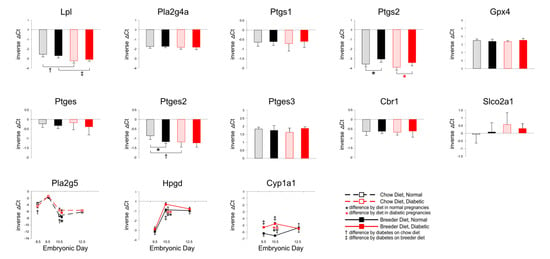
Figure 4.
Expression of genes involved in prostaglandin synthesis and metabolism. Quantitative PCR measurements were performed at various time points, in dams that were either fed chow or breeder diet, and became diabetic in the week before, or were normoglycemic at the time of mating. Data in bar diagrams were obtained from placentae isolated at E10.5.
As prostanoids can act in pro- and anti-inflammatory roles, we also surveyed genes that are known to be involved in inflammation. We had showed previously that Ankrd2 [13], which modulates NF-κB inflammatory responses [58], is specifically expressed in the placental labyrinth. Here, we focused on Metalloproteases 13, 1a, and 8, and protease inhibitors Kazal-type serine peptidase inhibitor domain 1 (Kazald1) which also contains an Igf-binding domain, the Kazal-type serine peptidase inhibitor Spink8, Serine protease inhibitor 16 (Spi16) and Thrombospondin type 1 domain containing 4 (Thsd4). Involved in turnover of extracellular matrix [59], this group of molecules contribute to the regulation of cell adhesion and migration [60,61,62]. Additional candidate genes encode Rnase2a, which attracts macrophages [63], Pore-forming protein-like (Pfpl), which is proposed to enhance trophoblast invasiveness [64], and Cysteine-rich C-terminal protein 1 (Crct1), which has been implicated in apoptosis [65].
Figure 5 demonstrates that these genes were all affected by maternal diet and diabetes. Mmp13 levels were lower under diabetic conditions in chow-fed dams, but higher when breeder diet was consumed; by E12.5, diabetes was associated with lower-than-normal expression, particularly in dams fed breeder diet. An effect of diet on Mmp1a transcripts was only evident at E10.5, with antipodal effects of diabetes at various time points. In contrast, consistently higher expression of Mmp8 was seen under diabetic conditions at E9.5 and E10.5. Lower Cathepsin Q levels were detected at E10.5 in chow-fed diabetic dams, but not at other time points or metabolic conditions. Maternal diabetes and dietary regimen affected Kazald1 expression at various time points with both dietary regimen, but as for Mmp13 and Mmp1a, trajectories changed over time. Likewise, expression of Spink8 reacted to diabetes and diet, whereas Spi16 was influenced only by diet at E10.5 and E12.5, in both metabolic conditions. Thsd4 levels were higher in diabetic than normal placentae, and normalized by E12.5. Rnase2 and Pfpl expression differed by metabolic condition at E9.5 and E10.5, and at E10.5 and E12.5, respectively. Crct1 transcript levels were higher in diabetic conditions with both diets, and, again, the major differences were seen at E9.5 and E10.5, after which diet had a greater influence at E12.5. Ankrd2 exhibited lower expression in diabetic placentae of chow-fed animals; breeder diet only had an effect at E9.5. Overall, where expression levels showed the greatest differences, the diabetic condition was typically associated with increased gene expression, although this was reversed for Ctsq and Ankrd2, which displayed reduced expression in diabetic placentae. In addition, the most profound differences were again evident by E10.5, further highlighting that placental gene expression this stage appears to be particularly sensitive to dietary and metabolic conditions.
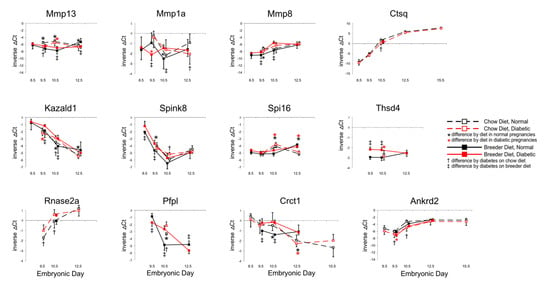
Figure 5.
Expression of genes involved in inflammation, cell adhesion and migration. Quantitative PCR measurements were performed at various time points, in dams that were either fed chow or breeder diet, and became diabetic in the week before, or were normoglycemic at the time of mating.
The complexity of gene-specific temporal trajectories, and of distinct responses to maternal diabetes and diet at different time points, raises the possibility that the underlying regulatory mechanisms could be highly specific to particular cell types in the placenta. The murine placenta is now known to harbor over 20 cell identities that are distinguishable on the basis of single-cell-based expression profiling [66,67]. Yet, the data currently available from such efforts are insufficient to allow unequivocal attribution of cell-type-specific gene expression (Salbaum and Kappen, unpublished observations). Based on conventional in situ hybridization methods, we previously demonstrated regionally restricted expression in the placenta for several genes in the diabetic pregnancy model, such as for Ascl2 and Ankrd2 in the labyrinth, Slc6a4 in giant cells, and Cyp1a1, Calcb and Cck in the decidual region of the placenta [13]; these studies also uncovered expression of Mmp9 in trophoblast giant cells, Thsd4 in giant cells, the labyrinth and decidua, and Sfrp5 and Rik9130008F23 exclusively in the decidual region. In the present study, we performed in situ hybridizations at E10.5 for 14 additional genes.
Figure 6 reveals that Lpl expression is strongest in the labyrinthine region, while Pla2g4 and Pla2g5 are predominantly expressed in the decidual region, with Pla2g4 also present in a layer of cells in the junctional zone in apposition to the labyrinth. The junctional zone is where Hpgd transcripts are also found, although positive cells appear in the decidual area in diabetic placentae, too. Signal for the prostaglandin transporter Slco2a1, which facilitates prostaglandin degradation, is detected throughout the decidual region, and also in the junctional zone, but entirely absent from the labyrinth. These patterns are consistent with the interpretation that arachidonic acid metabolites may be preferentially generated in the maternal part of the placenta, and that prostaglandin availability may also be regulated there, as well as in the junctional zone. Taken together with our quantitative assays indicating potentially elevated elimination of prostanoids, these results suggest that the reported deficiencies of arachidonic acid and prostaglandin E2 in diabetic pregnancies [68] could have their origin in the decidua and junctional zone, and that embryonic defects arising from deficiency of AA and PGE2, or both, are the result of perturbed paracrine mechanisms in decidua and placenta. Furthermore, as reduced availability of Prostaglandin E2 has been shown to impair migration of extravillous human first-trimester trophoblast cells [69], this would provide a mechanistic explanation for our earlier observations of reduced migration of spongiotrophoblast precursors to their destination in the junctional zone and their ectopic accumulation in the labyrinth.
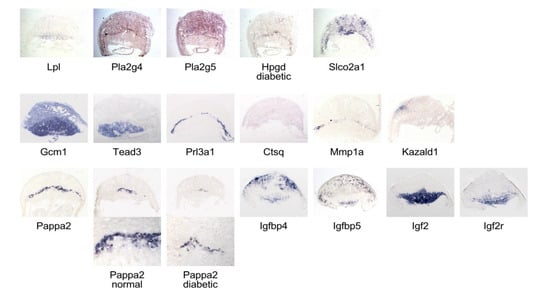
Figure 6.
In situ hybridization reveals region- and cell-type-specific gene expression in normal and diabetic placentae. Sections were produced from OCT-embedded frozen tissue from normoglycemic dams, except where indicated that the sample came from a diabetic female.
The labyrinth exhibits strong expression of the transcription factor Glial cells missing 1 (Gcm1), as reported previously [70] and of Transcription enhancer factor TEAD3 [71]. Crct1 and Pfpl transcripts were detected only in very few cells along the maternal-embryonic interface (not shown). Trophoblast giant cells express Prolactin Prl3d1 [72], and some of them display rather faint signals for Cathepsin Q mRNA. Mmp1a expression is also seen in the junctional zone, but protease inhibitor Kazald1 is only expressed in the decidual region, and at very low levels in labyrinth.
The junctional zone is the only area displaying strong signals for Pappa2, which were weaker and present in fewer cells in diabetic placenta at E10.5. This indicates that fewer spongiotrophoblasts were present already at this time point, foreshadowing the decreased thickness of the junctional zone we previously documented at E15.5 and E18.5 in diabetic placentae. The Igf binding protein transcripts Igfbp4 and Igfbp5 are expressed associated with maternal vessels in the decidual region, and also in the lower labyrinth. The labyrinth and spongiotrophoblasts strongly express Igf2, consistent with prior reports [73]; Igf2 receptor is also expressed in those areas. These results show that Igf-binding proteins are present in both maternal and embryonic compartments of the placenta, whereas Pappa2, which also controls Igf availability, is exclusive to the junctional zone. Relevant to diabetic pregnancies, reduced availability of Igf2, associated with reduced Pappa2 expression (also see Figure 1) is predicted to result in smaller placentae, and also reduced embryo growth [74,75]. Lower concentrations of free Igf2 have been linked to decreased placental nutrient transport [73,76], which, subsequently, may contribute to cause the fetal growth reduction we documented previously. Thus, our identification of Pappa2 as a gene whose expression is decreased by maternal diabetes in the placenta implicates impaired Igf2 signaling as the underlying mechanism for intrauterine growth restriction in type I diabetic mouse pregnancies.
In summary, this study investigated potential candidate genes for growth and cellular abnormalities in placentae of mice with type I diabetes during pregnancy. Our results allow us to attribute the growth deficiencies of placentae and embryos/fetuses in those pregnancies to impaired Igf2 signaling, with possible contribution of impaired Serotonin signaling. Furthermore, we provide evidence to link impaired cell migration to abnormal prostaglandin signaling. Notwithstanding the complex responsiveness of individual genes to maternal diabetes, maternal diet, or both, an overarching theme emerging from our studies is that gene expression in the midgestation placenta (E10.5) is particularly sensitive to metabolic and nutritional perturbations, possibly related to the switch from histiotrophic to hemotrophic nutrition. The molecular mechanisms by which maternal diabetes changes transcription of placental genes to cause intrauterine restriction of placental and fetal growth in the mouse, remain to be characterized.
Finally, potential implications for human pregnancies affected by type I diabetes deserve consideration: despite some structural and cellular distinctions, the process of placenta formation is highly similar between mouse and human, employing the same transcriptional networks and molecular pathways [77]. Under conditions of type I diabetes, the elevated risk for birth defects is also shared between both species [3]. However, two major differences exist between mouse and human type I diabetic pregnancies: (i) blood glucose levels in such pregnancies are very high in mouse models [11,12], but controllable to considerably lower levels in humans [78]. (ii) The longer pregnancy in humans is associated with a longer period of fetal growth in utero, while the mouse is born comparatively immaturely [79], limiting direct comparisons of gene expression data to early stages of pregnancy. Indeed, our results highlight and are consistent with prior reports that sensitivity of placental gene expression to perturbations is particularly pronounced during early stages of placenta development [77]. implicating potential vulnerabilities at comparable stages in human pregnancies.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/genes13010130/s1, File S1: Manufacturer’s datasheets for composition of LabDiet Purina 5001 and LabDiet Purina 5015, as downloaded from https://www.labdiet.com/Products/StandardDiets/index.html accessed on 31 October 2021.
Author Contributions
Conceptualization, C.K. (Claudia Kappen); methodology, C.K. (Claudia Kruger) and J.M.S.; software, J.M.S.; formal analysis, C.K. (Claudia Kappen) and C.K. (Claudia Kruger); data curation, C.K. (Claudia Kruger); writing—original draft preparation, C.K. (Claudia Kappen); writing—review and editing, C.K. (Claudia Kruger) and J.M.S.; visualization, C.K. (Claudia Kappen); funding acquisition, C.K. (Claudia Kappen) and J.M.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded in parts by grants from the Eunice Kennedy Shriver National Institute of Child Health and Human Development to C. Kappen (R01-HD037804 and R01-HD086604) and J.M. Salbaum (R01-HD085017), as well as through the Peggy M. Pennington Cole Endowed Chair in Developmental Biology at Pennington Biomedical Research Center (to C. Kappen).
Institutional Review Board Statement
Separate approval for the present study was not required because only archival samples were used. The approval information for the original sample collection is published [12,13].
Data Availability Statement
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request, in compliance with institutional policies of Pennington Biomedical Research Center/Louisiana State University System.
Acknowledgments
We thank Jackie MacGowan and Jennifer Martin for technical support with animal husbandry, sectioning and the in situ hybridization assays.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Mills, J.L. Malformations in infants of diabetic mothers. Teratology 1982, 25, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.M.; Tygstrup, I.; Pedersen, J. Congenital malformations in newborn infants of diabetic women: Correlation with maternal diabetic vascular complications. Lancet 1964, 1, 1124–1126. [Google Scholar] [CrossRef]
- Ornoy, A.; Reece, E.A.; Pavlinkova, G.; Kappen, C.; Miller, R.K. Effect of maternal diabetes on the embryo, fetus, and children: Congenital anomalies, genetic and epigenetic changes and developmental outcomes. Birth Defects Res. Part. C Embryo Today Rev. 2015, 105, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Kucera, J. Rate and type of congenital anomalies among offspring of diabetic women. J. Reprod Med. 1971, 7, 73–82. [Google Scholar] [PubMed]
- Vlachova, Z.; Bytoft, B.; Knorr, S.; Clausen, T.D.; Jensen, R.B.; Mathiesen, E.R.; Hojlund, K.; Ovesen, P.; Beck-Nielsen, H.; Gravholt, C.H.; et al. Increased metabolic risk in adolescent offspring of mothers with type 1 diabetes: The EPICOM study. Diabetologia 2015, 58, 14541463. [Google Scholar] [CrossRef]
- Plagemann, A.; Harder, T.; Kohlhoff, R.; Rohde, W.; Dorner, G. Overweight and obesity in infants of mothers with long-term insulin-dependent diabetes or gestational diabetes. Int. J. Obes. Relat. Metab. Disord. 1997, 21, 451–456. [Google Scholar] [CrossRef]
- Bozkurt, L.; Gobl, C.S.; Rami-Merhar, B.; Winhofer, Y.; Baumgartner-Parzer, S.; Schober, E.; Kautzky-Willer, A. The Cross-Link between Adipokines, Insulin Resistance and Obesity in Offspring of Diabetic Pregnancies. Horm. Res. Paediatr. 2016, 86, 300–308. [Google Scholar] [CrossRef]
- Dorner, G.; Plagemann, A. Perinatal hyperinsulinism as possible predisposing factor for diabetes mellitus, obesity and enhanced cardiovascular risk in later life. Horm. Metab. Res. 1994, 26, 213–221. [Google Scholar] [CrossRef]
- Catalano, P.M.; Hauguel-De Mouzon, S. Is it time to revisit the Pedersen hypothesis in the face of the obesity epidemic? Am. J. Obstet. Gynecol. 2011, 204, 479–487. [Google Scholar] [CrossRef]
- Burton, G.J.; Fowden, A.L.; Thornburg, K.L. Placental Origins of Chronic Disease. Physiol. Rev. 2016, 96, 1509–1565. [Google Scholar] [CrossRef]
- Kappen, C.; Kruger, C.; MacGowan, J.; Salbaum, J.M. Maternal diet modulates the risk for neural tube defects in a mouse model of diabetic pregnancy. Reprod. Toxicol. 2011, 31, 41–49. [Google Scholar] [CrossRef]
- Kappen, C.; Kruger, C.; MacGowan, J.; Salbaum, J.M. Maternal diet modulates placenta growth and gene expression in a mouse model of diabetic pregnancy. PLoS ONE 2012, 7, e38445. [Google Scholar] [CrossRef] [PubMed]
- Salbaum, J.M.; Kruger, C.; Zhang, X.; Delahaye, N.A.; Pavlinkova, G.; Burk, D.H.; Kappen, C. Altered gene expression and spongiotrophoblast differentiation in placenta from a mouse model of diabetes in pregnancy. Diabetologia 2011, 54, 1909–1920. [Google Scholar] [CrossRef]
- Inagaki, M.; Nishimura, T.; Nakanishi, T.; Shimada, H.; Noguchi, S.; Akanuma, S.I.; Tachikawa, M.; Hosoya, K.I.; Tamai, I.; Nakashima, E.; et al. Contribution of Prostaglandin Transporter OATP2A1/SLCO2A1 to Placenta-to-Maternal Hormone Signaling and Labor Induction. iScience 2020, 23, 101098. [Google Scholar] [CrossRef] [PubMed]
- Nicola, C.; Lala, P.K.; Chakraborty, C. Prostaglandin E2-mediated migration of human trophoblast requires RAC1 and CDC42. Biol. Reprod. 2008, 78, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Horita, H.; Kuroda, E.; Hachisuga, T.; Kashimura, M.; Yamashita, U. Induction of prostaglandin E2 production by leukemia inhibitory factor promotes migration of first trimester extravillous trophoblast cell line, HTR-8/SVneo. Hum. Reprod. 2007, 22, 1801–1809. [Google Scholar] [CrossRef]
- Kruger, C.; Talmadge, C.; Kappen, C. Expression of folate pathway genes in the cartilage of Hoxd4 and Hoxc8 transgenic mice. Birth Defects Res. A Clin. Mol. Teratol. 2006, 76, 216–229. [Google Scholar] [CrossRef]
- Kappen, C.; Kruger, C.; Jones, S.; Herion, N.J.; Salbaum, J.M. Maternal diet modulates placental nutrient transporter gene expression in a mouse model of diabetic pregnancy. PLoS ONE 2019, 14, e0224754. [Google Scholar] [CrossRef] [PubMed]
- Fowden, A.L. The insulin-like growth factors and feto-placental growth. Placenta 2003, 24, 803–812. [Google Scholar] [CrossRef]
- Wang, J.; Qiu, Q.; Haider, M.; Bell, M.; Gruslin, A.; Christians, J.K. Expression of pregnancy-associated plasma protein A2 during pregnancy in human and mouse. J. Endocrinol. 2009, 202, 337–345. [Google Scholar] [CrossRef]
- Gyrup, C.; Oxvig, C. Quantitative analysis of insulin-like growth factor-modulated proteolysis of insulin-like growth factor binding protein-4 and -5 by pregnancy-associated plasma protein-A. Biochemistry 2007, 46, 1972–1980. [Google Scholar] [CrossRef]
- Gaidamauskas, E.; Gyrup, C.; Boldt, H.B.; Schack, V.R.; Overgaard, M.T.; Laursen, L.S.; Oxvig, C. IGF dependent modulation of IGF binding protein (IGFBP) proteolysis by pregnancy-associated plasma protein-A (PAPP-A): Multiple PAPP-A-IGFBP interaction sites. Biochim. Biophys. Acta 2013, 1830, 2701–2709. [Google Scholar] [CrossRef]
- Conover, C.A.; Bale, L.K.; Overgaard, M.T.; Johnstone, E.W.; Laursen, U.H.; Fuchtbauer, E.M.; Oxvig, C.; van Deursen, J. Metalloproteinase pregnancy-associated plasma protein A is a critical growth regulatory factor during fetal development. Development 2004, 131, 1187–1194. [Google Scholar] [CrossRef]
- Christians, J.K.; de Zwaan, D.R.; Fung, S.H. Pregnancy associated plasma protein A2 (PAPP-A2) affects bone size and shape and contributes to natural variation in postnatal growth in mice. PLoS ONE 2013, 8, e56260. [Google Scholar] [CrossRef]
- Ozanne, S.E.; Nicholas Hales, C. Poor fetal growth followed by rapid postnatal catch-up growth leads to premature death. Mech. Ageing Dev. 2005, 126, 852–854. [Google Scholar] [CrossRef]
- Berends, L.M.; Dearden, L.; Tung, Y.C.L.; Voshol, P.; Fernandez-Twinn, D.S.; Ozanne, S.E. Programming of central and peripheral insulin resistance by low birthweight and postnatal catch-up growth in male mice. Diabetologia 2018, 61, 2225–2234. [Google Scholar] [CrossRef]
- Essalmani, R.; Hamelin, J.; Marcinkiewicz, J.; Chamberland, A.; Mbikay, M.; Chretien, M.; Seidah, N.G.; Prat, A. Deletion of the gene encoding proprotein convertase 5/6 causes early embryonic lethality in the mouse. Mol. Cell. Biol. 2006, 26, 354–361. [Google Scholar] [CrossRef]
- Zhang, H.; Bradley, A. Mice deficient for BMP2 are nonviable and have defects in amnion/chorion and cardiac development. Development 1996, 122, 2977–2986. [Google Scholar] [CrossRef]
- Li, Y.; Klena, N.T.; Gabriel, G.C.; Liu, X.; Kim, A.J.; Lemke, K.; Chen, Y.; Chatterjee, B.; Devine, W.; Damerla, R.R.; et al. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature 2015, 521, 520–524. [Google Scholar] [CrossRef]
- Koscielny, G.; Yaikhom, G.; Iyer, V.; Meehan, T.F.; Morgan, H.; Atienza-Herrero, J.; Blake, A.; Chen, C.K.; Easty, R.; Di Fenza, A.; et al. The International Mouse Phenotyping Consortium Web Portal, a unified point of access for knockout mice and related phenotyping data. Nucleic Acids Res. 2014, 42, D802–D809. [Google Scholar] [CrossRef]
- Holmbeck, K.; Bianco, P.; Caterina, J.; Yamada, S.; Kromer, M.; Kuznetsov, S.A.; Mankani, M.; Robey, P.G.; Poole, A.R.; Pidoux, I.; et al. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell 1999, 99, 81–92. [Google Scholar] [CrossRef]
- Zhang, Y.; Wen, G.; Shao, G.; Wang, C.; Lin, C.; Fang, H.; Balajee, A.S.; Bhagat, G.; Hei, T.K.; Zhao, Y. TGFBI deficiency predisposes mice to spontaneous tumor development. Cancer Res. 2009, 69, 37–44. [Google Scholar] [CrossRef]
- Boing, M.; Brand-Saberi, B.; Napirei, M. Murine transcription factor Math6 is a regulator of placenta development. Sci. Rep. 2018, 8, 14997. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garcia, V.; Fineberg Em Wilson, R.; Murray, A.; Mazzeo, C.I.; Tudor, C.; Sienerth, A.; White, J.K.; Tuck, E.; Ryder, E.J. Placentation defects are highly prevalent in embryonic lethal mouse mutants. Nature 2018, 555, 463–468. [Google Scholar] [CrossRef]
- Guillemot, F.; Nagy, A.; Auerbach, A.; Rossant, J.; Joyner, A.L. Essential role of Mash-2 in extraembryonic development. Nature 1994, 371, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Yavarone, M.S.; Shuey, D.L.; Sadler, T.W.; Lauder, J.M. Serotonin uptake in the ectoplacental cone and placenta of the mouse. Placenta 1993, 14, 149–161. [Google Scholar] [CrossRef]
- Lescisin, K.R.; SVarmuza Rossant, J. Isolation and characterization of a novel trophoblast-specific cDNA in the mouse. Genes Dev. 1988, 2, 1639–1646. [Google Scholar] [CrossRef]
- Wiemers, D.O.; Shao, L.J.; Ain, R.; Dai, G.; Soares, M.J. The mouse prolactin gene family locus. Endocrinology 2003, 144, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Shuey, D.L.; Yavarone, M.; Sadler, T.W.; Lauder, J.M. Serotonin and morphogenesis in the cultured mouse embryo. Adv. Exp. Med. Biol. 1990, 265, 205–215. [Google Scholar]
- Lauder, J.M.; JAWallace Krebs, H. Roles for serotonin in neuroembryogenesis. Adv. Exp. Med. Biol. 1981, 133, 477–506. [Google Scholar]
- Murphy, D.L.; Fox, M.A.; Timpano, K.R.; Moya, P.R.; Ren-Patterson, R.; Andrews, A.M.; Holmes, A.; Lesch, K.P.; Wendland, J.R. How the serotonin story is being rewritten by new gene-based discoveries principally related to SLC6A4, the serotonin transporter gene, which functions to influence all cellular serotonin systems. Neuropharmacology 2008, 55, 932–960. [Google Scholar] [CrossRef]
- Cote, F.; Fligny, C.; Bayard, E.; Launay, J.M.; Gershon, M.D.; Mallet, J.; Vodjdani, G. Maternal serotonin is crucial for murine embryonic development. Proc. Natl. Acad. Sci. USA 2007, 104, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, C.S. Placental serotonin signaling, pregnancy outcomes, and regulation of fetal brain development. Biol. Reprod. 2020, 102, 532–538. [Google Scholar] [CrossRef]
- McGrath, K.E.; Koniski, A.D.; Malik, J.; Palis, J. Circulation is established in a stepwise pattern in the mammalian embryo. Blood 2003, 101, 1669–1676. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, M.; Nishimura, T.; Akanuma, S.I.; Nakanishi, T.; Tachikawa, M.; Tamai, I.; Hosoya, K.I.; Nakashima, E.; Tomi, M. Co-localization of microsomal prostaglandin E synthase-1 with cyclooxygenase-1 in layer II of murine placental syncytiotrophoblasts. Placenta 2017, 53, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Chida, S.; Mettler, L. Effects of indomethacin, prostaglandin E2 and prostaglandin F2 alpha on mouse blastocyst attachment and trophoblastic outgrowth in vitro. Prostaglandins 1989, 37, 411–416. [Google Scholar] [CrossRef]
- Holmes, P.V.; Gordashko, B.J. Evidence of prostaglandin involvement in blastocyst implantation. J. Embryol. Exp. Morphol. 1980, 55, 109–122. [Google Scholar] [CrossRef]
- Song, H.; Lim, H.; Paria, B.C.; Matsumoto, H.; Swift, L.L.; Morrow, J.; Bonventre, J.V.; Dey, S.K. Cytosolic phospholipase A2alpha is crucial [correction of A2alpha deficiency is crucial] for ’on-time’ embryo implantation that directs subsequent development. Development 2002, 129, 2879–2889. [Google Scholar] [CrossRef]
- Scherle, P.A.; Ma, W.; Lim, H.; Dey, S.K.; Trzaskos, J.M. Regulation of cyclooxygenase-2 induction in the mouse uterus during decidualization: An event of early pregnancy. J. Biol. Chem. 2000, 275, 37086–37092. [Google Scholar] [CrossRef]
- White, V.; Jawerbaum, A.; Sinner, D.; Pustovrh, C.; Capobianco, E.; Gonzalez, E. Oxidative stress and altered prostanoid production in the placenta of streptozotocin-induced diabetic rats. Reprod. Fertil. Dev. 2002, 14, 117–123. [Google Scholar] [CrossRef]
- Zhao, J.; Del Bigio, M.R.; Weiler, H.A. Maternal arachidonic acid supplementation improves neurodevelopment of offspring from healthy and diabetic rats. Prostaglandins Leukot Essent Fatty Acids 2009, 81, 349–356. [Google Scholar] [CrossRef]
- Goto, M.P.; Goldman ASUhing, M.R. PGE2 prevents anomalies induced by hyperglycemia or diabetic serum in mouse embryos. Diabetes 1992, 41, 1644–1650. [Google Scholar] [CrossRef] [PubMed]
- Reece, E.A.; Homko, C.J.; Wu, Y.K.; Wiznitzer, A. The role of free radicals and membrane lipids in diabetes-induced congenital malformations. J. Soc. Gynecol. Investig. 1998, 5, 178–187. [Google Scholar] [CrossRef]
- Baker, L.; Piddington, R.; Goldman, A.; Egler, J.; Moehring, J. Myo-inositol and prostaglandins reverse the glucose inhibition of neural tube fusion in cultured mouse embryos. Diabetologia 1990, 33, 593–596. [Google Scholar] [CrossRef]
- Goldman, A.S.; Baker, L.; Piddington, R.; Marx, B.; Herold, R.; Egler, J. Hyperglycemia-induced teratogenesis is mediated by a functional deficiency of arachidonic acid. Proc. Natl. Acad. Sci. USA 1985, 82, 8227–8231. [Google Scholar] [CrossRef]
- Maiorino, M.; MConrad Ursini, F. GPx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxid. Redox Signal. 2018, 29, 61–74. [Google Scholar] [CrossRef]
- Kroetz, D.L.; Xu, F. Regulation and inhibition of arachidonic acid omega-hydroxylases and 20-HETE formation. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 413–438. [Google Scholar] [CrossRef]
- Bean, C.; Verma, N.K.; Yamamoto, D.L.; Chemello, F.; Cenni, V.; Filomena, M.C.; Chen, J.; Bang, M.L.; Lanfranchi, G. Ankrd2 is a modulator of NF-kappaB-mediated inflammatory responses during muscle differentiation. Cell Death Dis. 2014, 5, e1002. [Google Scholar] [CrossRef]
- Manabe, R.; Tsutsui, K.; Yamada, T.; Kimura, M.; Nakano, I.; Shimono, C.; Sanzen, N.; Furutani, Y.; Fukuda, T.; Oguri, Y.; et al. Transcriptome-based systematic identification of extracellular matrix proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 12849–12854. [Google Scholar] [CrossRef]
- Zigrino, P.; Kuhn, I.; Bauerle, T.; Zamek, J.; Fox, J.W.; Neumann, S.; Licht, A.; Schorpp-Kistner, M.; Angel, P.; Mauch, C. Stromal expression of MMP-13 is required for melanoma invasion and metastasis. J. Investig. Dermatol. 2009, 129, 2686–2693. [Google Scholar] [CrossRef]
- Sun, J.; Ooms, L.; Bird, C.H.; Sutton, V.R.; Trapani, J.A.; Bird, P.I. A new family of 10 murine ovalbumin serpins includes two homologs of proteinase inhibitor 8 and two homologs of the granzyme B inhibitor (proteinase inhibitor 9). J. Biol. Chem. 1997, 272, 15434–15441. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, K.; Manabe, R.; Yamada, T.; Nakano, I.; Oguri, Y.; Keene, D.R.; Sengle, G.; Sakai, L.Y.; Sekiguchi, K. ADAMTSL-6 is a novel extracellular matrix protein that binds to fibrillin-1 and promotes fibrillin-1 fibril formation. J. Biol. Chem. 2010, 285, 4870–4882. [Google Scholar] [CrossRef]
- Yamada, K.J.; Barker, T.; Dyer, K.D.; Rice, T.A.; Percopo, C.M.; Garcia-Crespo, K.E.; Cho, S.; Lee, J.J.; Druey, K.M.; Rosenberg, H.F. Eosinophil-associated ribonuclease 11 is a macrophage chemoattractant. J. Biol. Chem. 2015, 290, 8863–8875. [Google Scholar] [CrossRef] [PubMed]
- Hemberger, M.; Himmelbauer, H.; Ruschmann, J.; Zeitz, C.; Fundele, R. cDNA subtraction cloning reveals novel genes whose temporal and spatial expression indicates association with trophoblast invasion. Dev. Biol. 2000, 222, 158–169. [Google Scholar] [CrossRef][Green Version]
- Wu, N.; Song, Y.; Pang, L.; Chen, Z. CRCT1 regulated by microRNA-520 g inhibits proliferation and induces apoptosis in esophageal squamous cell cancer. Tumour. Biol. 2016, 37, 8271–8279. [Google Scholar] [CrossRef]
- Marsh, B.; Blelloch, R. Single nuclei RNA-seq of mouse placental labyrinth development. Elife 2020, 9, e60266. [Google Scholar] [CrossRef]
- Nelson, A.C.; Mould, A.W.; Bikoff, E.K.; Robertson, E.J. Single-cell RNA-seq reveals cell type-specific transcriptional signatures at the maternal-foetal interface during pregnancy. Nat. Commun. 2016, 7, 11414. [Google Scholar] [CrossRef]
- Piddington, R.; Joyce, J.; Dhanasekaran, P.; Baker, L. Diabetes mellitus affects prostaglandin E2 levels in mouse embryos during neurulation. Diabetologia 1996, 39, 15–20. [Google Scholar] [CrossRef]
- Nicola, C.; Timoshenko, A.V.; Dixon, S.J.; Lala, P.K.; Chakraborty, C. EP1 receptor-mediated migration of the first trimester human extravillous trophoblast: The role of intracellular calcium and calpain. J. Clin. Endocrinol. Metab. 2005, 90, 4736–4746. [Google Scholar] [CrossRef] [PubMed]
- Anson-Cartwright, L.; Dawson, K.; Holmyard, D.; Fisher, S.J.; Lazzarini, R.A.; Cross, J.C. The glial cells missing-1 protein is essential for branching morphogenesis in the chorioallantoic placenta. Nat. Genet. 2000, 25, 311–314. [Google Scholar] [CrossRef]
- Jacquemin, P.; Sapin, V.; Alsat, E.; Evain-Brion, D.; Dolle, P.; Davidson, I. Differential expression of the TEF family of transcription factors in the murine placenta and during differentiation of primary human trophoblasts in vitro. Dev. Dyn. 1998, 212, 423–436. [Google Scholar] [CrossRef]
- Colosi, P.; Swiergiel, J.J.; Wilder, E.L.; Oviedo, A.; Linzer, D.I. Characterization of proliferin-related protein. Mol. Endocrinol. 1988, 2, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Constancia, M.; Angiolini, E.; Sandovici, I.; Smith, P.; Smith, R.; Kelsey, G.; Dean, W.; Ferguson-Smith, A.; Sibley, C.P.; Reik, W.; et al. Adaptation of nutrient supply to fetal demand in the mouse involves interaction between the Igf2 gene and placental transporter systems. Proc. Natl. Acad. Sci. USA 2005, 102, 19219–19224. [Google Scholar] [CrossRef] [PubMed]
- Aykroyd, B.R.L.; SJTunster Sferruzzi-Perri, A.N. Igf2 deletion alters mouse placenta endocrine capacity in a sexually dimorphic manner. J. Endocrinol. 2020, 246, 93–108. [Google Scholar] [CrossRef]
- Constancia, M.; Hemberger, M.; Hughes, J.; Dean, W.; Ferguson-Smith, A.; Fundele, R.; Stewart, F.; Kelsey, G.; Fowden, A.; Sibley, C.; et al. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature 2002, 417, 945–948. [Google Scholar] [CrossRef]
- Sferruzzi-Perri, A.N.; Vaughan, O.R.; Coan, P.M.; Suciu, M.C.; Darbyshire, R.; Constancia, M.; Burton, G.J.; Fowden, A.L. Placental-specific Igf2 deficiency alters developmental adaptations to undernutrition in mice. Endocrinology 2011, 152, 3202–3212. [Google Scholar] [CrossRef] [PubMed]
- Hemberger, M.; Hanna, C.W.; Dean, W. Mechanisms of early placental development in mouse and humans. Nat. Rev. Genet. 2020, 21, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, P.A.; Frias, J.P.; Peters, K.A.; Chillara, B.; Garg, S.K. Optimizing insulin therapy in pregnant women with type 1 diabetes mellitus. Treat Endocrinol. 2002, 1, 235–240. [Google Scholar] [CrossRef]
- Hogan, B.; Beddington, R.; Costantini, F.; Lacy, E. Manipulating the Mouse Embryo: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1994. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).