
Analysis of Simian Endogenous Retrovirus (SERV) Full-Length Proviruses in Old World Monkey Genomes


Abstract
1. Introduction
2. Materials and Methods
2.1. Identifying Simian Endogenous Retrovirus (SERV) Sequences in Cercopithecinae
2.2. Analysis of Proviral Sequences
3. Results
- Sequence and assembly quality may vary;
- Not all species have been sequenced;
- For most species, only a single genome assembly is available;
- Y chromosome sequences are not always present;
- Heterozygous EVEs are omitted from assemblies;
- Full-length proviruses may be younger than fragmented ones;
- It is not likely that all viral variation has been captured in the germ line.
3.1. Phylogenetic Analysis of Cercopithecinae SERV Proviruses
3.1.1. SERV Diversity in OWM
3.1.2. Cer-SERV Integrations in the T. gelada Genome
3.2. Mutation Patterns in SERV Proviruses
3.2.1. CpG Methylation-Associated Mutations
3.2.2. APOBEC3G and -Non-3G Signature Patterns
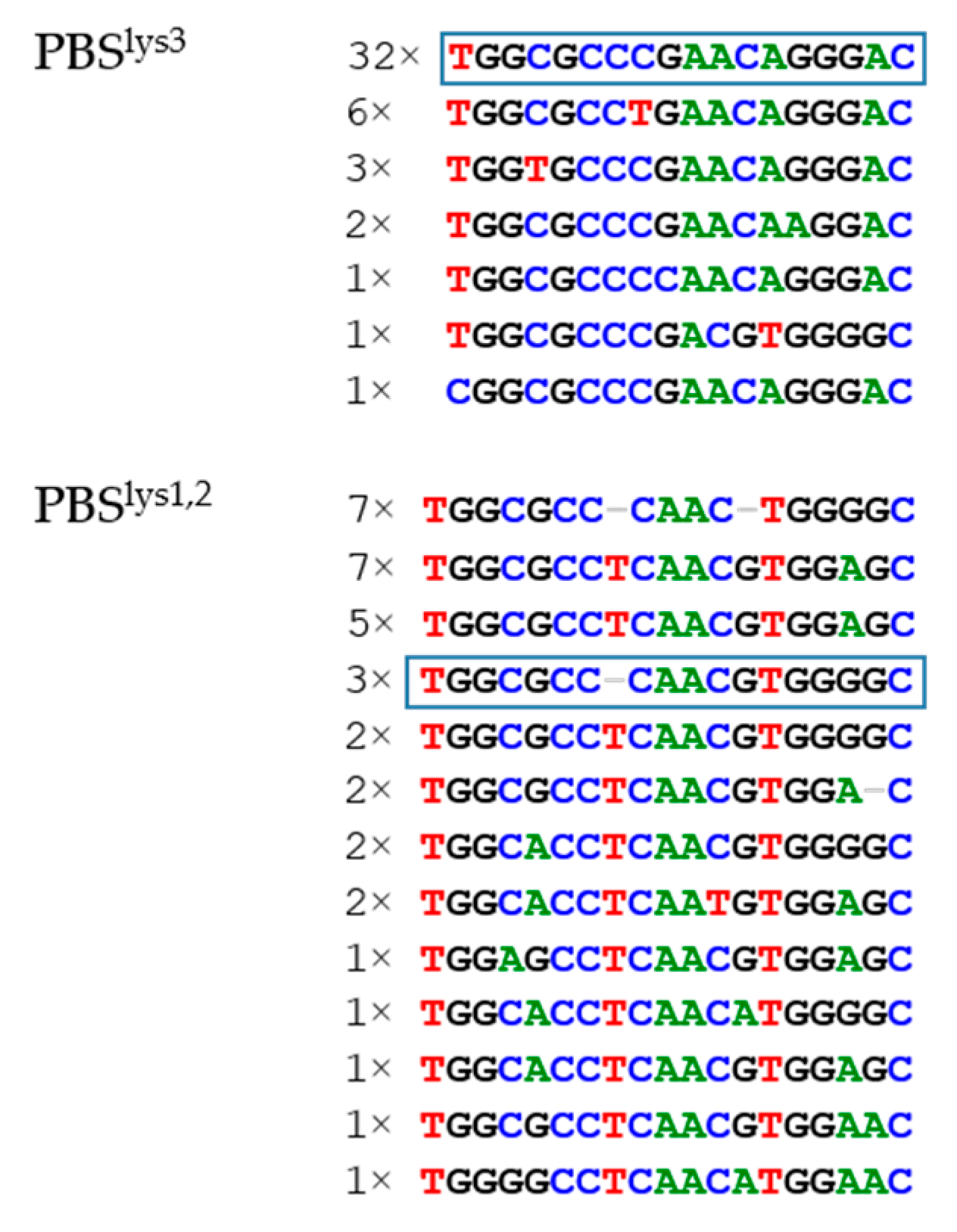
3.2.3. PBS Variation

3.3. Genomic Features of SERV Proviruses
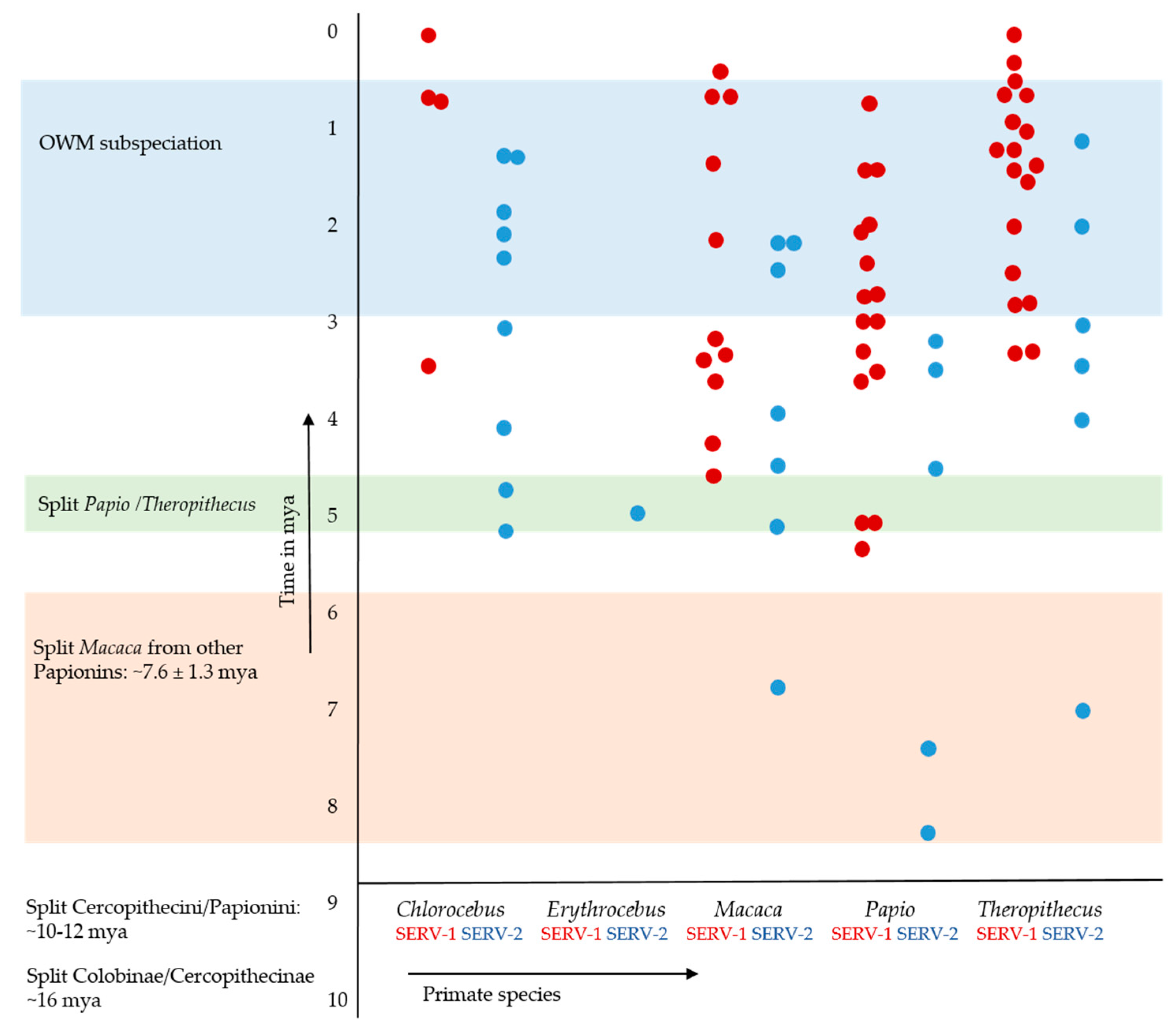
3.3.1. Cer-SERV LTR Sequence Diversity and Dating
3.3.2. Cer-SERV Coding Region Diversity
3.4. Shared SERV Proviral Integrations in Macaques
3.5. Shared Cer-SERV Proviral Integrations in Baboon and Gelada
4. Discussion
5. Conclusions
- Shared, ancient proviral Cer-SERV integrations between species are rare;
- Most Cer-SERV proviral integrations are relatively young;
- Species-specific young, but not old, Cer-SERV clusters are seen in OWM genomes;
- Cer-SERV PBS sequences show evidence of tRF suppression, a mechanism only operational in the early embryo;
- Some young Cer-SERV-1 LTRs have acquired binding sites for embryo-specific transcription factors;
- Cer-SERV pol genes are more often uninterrupted than Cer-SERV env genes;
- Cer-SERV Env expression from integrated proviruses should be able to block the receptor to prevent Env-mediated reinfection of the cell. However, reinfections of the germ line are common.
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chiu, E.S.; VandeWoude, S. Endogenous retroviruses drive resistance and promotion of exogenous retroviral homologs. Annu. Rev. Anim. Biosci. 2021, 9, 225–248. [Google Scholar] [CrossRef] [PubMed]
- van der Kuyl, A.C.; Berkhout, B. Viruses in the reproductive tract: On their way to the germ line? Virus Res. 2020, 286, 198101. [Google Scholar] [CrossRef]
- van der Kuyl, A.C.; Mang, R.; Dekker, J.T.; Goudsmit, J. Complete nucleotide sequence of simian endogenous type D retrovirus with intact genome organization: Evidence for ancestry to simian retrovirus and baboon endogenous virus. J. Virol. 1997, 71, 3666–3676. [Google Scholar] [CrossRef] [PubMed]
- van der Kuyl, A.C. Contemporary distribution, estimated age, and prehistoric migrations of Old World monkey retroviruses. Epidemiologia 2021, 2, 46–67. [Google Scholar] [CrossRef]
- Power, M.D.; Marx, P.A.; Bryant, M.L.; Gardner, M.B.; Barr, P.J.; Luciw, P.A. Nucleotide sequence of SRV-1, a type D simian acquired immune deficiency syndrome retrovirus. Science 1986, 231, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, C.; Sekizuka, T.; Kuroda, M.; Kasai, F.; Saito, K.; Ikeda, M.; Yamaji, T.; Osada, N.; Hanada, K. Novel endogenous simian retroviral integrations in Vero cells: Implications for quality control of a human vaccine cell substrate. Sci. Rep. 2018, 8, 644. [Google Scholar] [CrossRef]
- Ikeda, M.; Satomura, K.; Sekizuka, T.; Hanada, K.; Endo, T.; Osada, N. Comprehensive phylogenomic analysis reveals a novel cluster of simian endogenous retroviral sequences in Colobinae monkeys. Am. J. Primatol. 2018, 80, e22882. [Google Scholar] [CrossRef] [PubMed]
- Elton, S. Environmental correlates of the cercopithecoid radiations. Folia Primatol. 2007, 78, 344–364. [Google Scholar] [CrossRef]
- Schorn, A.J.; Gutbrod, M.J.; LeBlanc, C.; Martienssen, R. LTR-Retrotransposon Control by tRNA-Derived Small RNAs. Cell 2017, 170, 61–71.e11. [Google Scholar] [CrossRef] [PubMed]
- Batra, S.S.; Levy-Sakin, M.; Robinson, J.; Guillory, J.; Durinck, S.; Vilgalys, T.P.; Kwok, P.Y.; Cox, L.A.; Seshagiri, S.; Song, Y.S.; et al. Accurate assembly of the olive baboon (Papio anubis) genome using long-read and Hi-C data. Gigascience 2020, 9, giaa134. [Google Scholar] [CrossRef]
- Rogers, J.; Raveendran, M.; Harris, R.A.; Mailund, T.; Leppälä, K.; Athanasiadis, G.; Schierup, M.H.; Cheng, J.; Munch, K.; Walker, J.A.; et al. The comparative genomics and complex population history of Papio baboons. Sci. Adv. 2019, 5, eaau6947. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Kishino, H.; Yano, T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Siepel, A.C.; Halpern, A.L.; Macken, C.; Korber, B.T. A computer program designed to screen rapidly for HIV type 1 intersubtype recombinant sequences. AIDS Res. Hum. Retrovir. 1995, 11, 1413–1416. [Google Scholar] [CrossRef]
- Rose, P.P.; Korber, B.T. Detecting hypermutations in viral sequences with an emphasis on G --> A hypermutation. Bioinformatics 2000, 16, 400–401. [Google Scholar] [CrossRef] [PubMed]
- Alinejad-Rokny, H.; Ebrahimi, D. A method to avoid errors associated with the analysis of hypermutated viral sequences by alignment-based methods. J. Biomed. Inf. 2015, 58, 220–225. [Google Scholar] [CrossRef]
- Osada, N.; Kohara, A.; Yamaji, T.; Hirayama, N.; Kasai, F.; Sekizuka, T.; Kuroda, M.; Hanada, K. The genome landscape of the african green monkey kidney-derived vero cell line. DNA Res. 2014, 21, 673–683. [Google Scholar] [CrossRef]
- Sonigo, P.; Barker, C.; Hunter, E.; Wain-Hobson, S. Nucleotide sequence of Mason-Pfizer monkey virus: An immunosuppressive D-type retrovirus. Cell 1986, 45, 375–385. [Google Scholar] [CrossRef]
- Bronson, E.C.; Anderson, J.N. Nucleotide composition as a driving force in the evolution of retroviruses. J. Mol. Evol. 1994, 38, 506–532. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Pontiller, J.; Mauceli, E.; Ernst, W.; Baumann, M.; Biagi, T.; Swofford, R.; Russell, P.; Zody, M.C.; Di Palma, F.; et al. Exploiting nucleotide composition to engineer promoters. PLoS ONE 2011, 6, e20136. [Google Scholar] [CrossRef]
- Conley, A.B.; Jordan, I.K. Cell type-specific termination of transcription by transposable element sequences. Mob. DNA 2012, 3, 15. [Google Scholar] [CrossRef]
- Rotman, G.; Itin, A.; Keshet, E. ‘Solo’ large terminal repeats (LTR) of an endogenous retrovirus-like gene family (VL30) in the mouse genome. Nucleic Acids Res. 1984, 12, 2273–2282. [Google Scholar] [CrossRef][Green Version]
- Yang, A.S.; Gonzalgo, M.L.; Zingg, J.M.; Millar, R.P.; Buckley, J.D.; Jones, P.A. The rate of CpG mutation in Alu repetitive elements within the p53 tumor suppressor gene in the primate germline. J. Mol. Biol. 1996, 258, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Nachman, M.W.; Crowell, S.L. Estimate of the mutation rate per nucleotide in humans. Genetics 2000, 156, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Lian, X.; Gao, C.; Sun, X.; Einkauf, K.B.; Chevalier, J.M.; Chen, S.M.Y.; Hua, S.; Rhee, B.; Chang, K.; et al. Distinct viral reservoirs in individuals with spontaneous control of HIV-1. Nature 2020, 585, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.A.; Wiegand, H.L.; Moore, M.D.; Schäfer, A.; McClure, M.O.; Cullen, B.R. Foamy virus Bet proteins function as novel inhibitors of the APOBEC3 family of innate antiretroviral defense factors. J. Virol. 2005, 79, 8724–8731. [Google Scholar] [CrossRef] [PubMed]
- Doehle, B.P.; Bogerd, H.P.; Wiegand, H.L.; Jouvenet, N.; Bieniasz, P.D.; Hunter, E.; Cullen, B.R. The betaretrovirus Mason-Pfizer monkey virus selectively excludes simian APOBEC3G from virion particles. J. Virol. 2006, 80, 12102–12108. [Google Scholar] [CrossRef][Green Version]
- Chiu, Y.L.; Greene, W.C. The APOBEC3 cytidine deaminases: An innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu. Rev. Immunol. 2008, 26, 317–353. [Google Scholar] [CrossRef]
- Lee, Y.N.; Bieniasz, P.D. Reconstitution of an infectious human endogenous retrovirus. PLoS Pathog. 2007, 3, e10. [Google Scholar] [CrossRef]
- Armitage, A.E.; Katzourakis, A.; de Oliveira, T.; Welch, J.J.; Belshaw, R.; Bishop, K.N.; Kramer, B.; McMichael, A.J.; Rambaut, A.; Iversen, A.K. Conserved footprints of APOBEC3G on hypermutated human immunodeficiency virus type 1 and human endogenous retrovirus HERV-K(HML2) sequences. J. Virol. 2008, 82, 8743–8761. [Google Scholar] [CrossRef]
- Lee, Y.N.; Malim, M.H.; Bieniasz, P.D. Hypermutation of an ancient human retrovirus by APOBEC3G. J. Virol. 2008, 82, 8762–8770. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.L.; Emerman, M.; Malik, H.S. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2004, 2, e275. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Webb, D.M. Rapid evolution of primate antiviral enzyme APOBEC3G. Hum. Mol. Genet. 2004, 13, 1785–1791. [Google Scholar] [CrossRef]
- Itin, A.; Keshet, E. Primer binding sites corresponding to several tRNA species are present in DNAs of different members of the same retrovirus-like gene family (VL30). J. Virol. 1985, 54, 236–239. [Google Scholar] [CrossRef]
- Das, A.T.; Klaver, B.; Berkhout, B. Sequence variation of the human immunodeficiency virus primer-binding site suggests the use of an alternative tRNA(Lys) molecule in reverse transcription. J. Gen. Virol. 1997, 78, 837–840. [Google Scholar] [CrossRef] [PubMed]
- Das, A.T.; Vink, M.; Berkhout, B. Alternative tRNA priming of human immunodeficiency virus type 1 reverse transcription explains sequence variation in the primer-binding site that has been attributed to APOBEC3G activity. J. Virol. 2005, 79, 3179–3181. [Google Scholar] [CrossRef]
- Fennessey, C.M.; Camus, C.; Immonen, T.T.; Reid, C.; Maldarelli, F.; Lifson, J.D.; Keele, B.F. Low-level alternative tRNA priming of reverse transcription of HIV-1 and SIV in vivo. Retrovirology 2019, 16, 11. [Google Scholar] [CrossRef]
- Cullen, H.; Schorn, A.J. Endogenous retroviruses walk a fine line between priming and silencing. Viruses 2020, 12, 792. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, W.; Cui, Y.C.; Pan, Q.; Zhu, W.; Gendron, P.; Guo, F.; Cen, S.; Witcher, M.; Liang, C. HIV-1 employs multiple mechanisms to resist Cas9/Single Guide RNA targeting the viral Primer Binding Site. J. Virol. 2018, 92, e01135-18. [Google Scholar] [CrossRef]
- Guo, Y.; Costa, R.; Ramsey, H.; Starnes, T.; Vance, G.; Robertson, K.; Kelley, M.; Reinbold, R.; Scholer, H.; Hromas, R. The embryonic stem cell transcription factors Oct-4 and FoxD3 interact to regulate endodermal-specific promoter expression. Proc. Natl. Acad. Sci. USA 2002, 99, 3663–3667. [Google Scholar] [CrossRef]
- Granadino, B.; Arias-de-la-Fuente, C.; Pérez-Sánchez, C.; Párraga, M.; López-Fernández, L.A.; del Mazo, J.; Rey-Campos, J. Fhx (Foxj2) expression is activated during spermatogenesis and very early in embryonic development. Mech. Dev. 2000, 97, 157–160. [Google Scholar] [CrossRef]
- Dangel, A.W.; Baker, B.J.; Mendoza, A.R.; Yu, C.Y. Complement component C4 gene intron 9 as a phylogenetic marker for primates: Long terminal repeats of the endogenous retrovirus ERV-K(C4) are a molecular clock of evolution. Immunogenetics 1995, 42, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E.; Coffin, J.M. Constructing primate phylogenies from ancient retrovirus sequences. Proc. Natl. Acad. Sci. USA 1999, 96, 10254–10260. [Google Scholar] [CrossRef]
- Zhuo, X.; Feschotte, C. Cross-species transmission and differential fate of an endogenous retrovirus in three mammal lineages. PLoS Pathog. 2015, 11, e1005279. [Google Scholar] [CrossRef] [PubMed]
- Roos, C.; Kothe, M.; Alba, D.M.; Delson, E.; Zinner, D. The radiation of macaques out of Africa: Evidence from mitogenome divergence times and the fossil record. J. Hum. Evol. 2019, 133, 114–132. [Google Scholar] [CrossRef] [PubMed]
- Tosi, A.J.; Morales, J.C.; Melnick, D.J. Paternal, maternal, and biparental molecular markers provide unique windows onto the evolutionary history of macaque monkeys. Evolution 2003, 57, 1419–1435. [Google Scholar] [CrossRef] [PubMed]
- Kanthaswamy, S.; Satkoski, J.; George, D.; Kou, A.; Erickson, B.J.; Smith, D.G. Interspecies hybridization and the stratification of nuclear genetic variation of rhesus (Macaca mulatta) and long-tailed macaques (Macaca fascicularis). Int. J. Primatol. 2008, 29, 1295–1311. [Google Scholar] [CrossRef]
- Marmi, J.; Bertranpetit, J.; Terradas, J.; Takenaka, O.; Domingo-Roura, X. Radiation and phylogeography in the Japanese macaque, Macaca fuscata. Mol. Phylogenet. Evol. 2004, 30, 676–685. [Google Scholar] [CrossRef]
- Chu, J.H.; Lin, Y.S.; Wu, H.Y. Evolution and dispersal of three closely related macaque species, Macaca mulatta, M. cyclopis, and M. fuscata, in the eastern Asia. Mol. Phylogenet. Evol. 2007, 43, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.E.; Disotell, T.R. Nuclear gene trees and the phylogenetic relationships of the mangabeys (Primates: Papionini). Mol. Biol. Evol. 1998, 15, 892–900. [Google Scholar] [CrossRef]
- Walker, J.A.; Jordan, V.E.; Storer, J.M.; Steely, C.J.; Gonzalez-Quiroga, P.; Beckstrom, T.O.; Rewerts, L.C.; St Romain, C.P.; Rockwell, C.E.; Rogers, J.; et al. Alu insertion polymorphisms shared by Papio baboons and Theropithecus gelada reveal an intertwined common ancestry. Mob. DNA 2019, 10, 46. [Google Scholar] [CrossRef]
- van der Kuyl, A.C.; Dekker, J.T. St. Kitts green monkeys originate from West Africa: Genetic evidence from feces. Am. J. Primatol. 1996, 40, 361–364. [Google Scholar] [CrossRef]
- Bhatt, S.; Katzourakis, A.; Pybus, O.G. Detecting natural selection in RNA virus populations using sequence summary statistics. Infect. Genet. Evol. 2010, 10, 421–430. [Google Scholar] [CrossRef][Green Version]
- Sun, M.A.; Wolf, G.; Wang, Y.; Senft, A.D.; Ralls, S.; Jin, J.; Dunn-Fletcher, C.E.; Muglia, L.J.; Macfarlan, T.S. Endogenous retroviruses drive lineage-specific regulatory evolution across primate and rodent placentae. Mol. Biol. Evol. 2021, 38, 4992–5004. [Google Scholar] [CrossRef]
- Senft, A.D.; Macfarlan, T.S. Transposable elements shape the evolution of mammalian development. Nat. Rev. Genet. 2021, 22, 691–711. [Google Scholar] [CrossRef] [PubMed]
- Magiorkinis, G.; Gifford, R.J.; Katzourakis, A.; De Ranter, J.; Belshaw, R. Env-less endogenous retroviruses are genomic superspreaders. Proc. Natl. Acad. Sci. USA 2012, 109, 7385–7390. [Google Scholar] [CrossRef] [PubMed]
- Tchenio, T.; Heidmann, T. Defective retroviruses can disperse in the human genome by intracellular transposition. J. Virol. 1991, 65, 2113–2118. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.; Johnson, W.E. Retroviruses of the RDR superinfection interference group: Ancient origins and broad host distribution of a promiscuous Env gene. Curr. Opin. Virol. 2017, 25, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E. Origins and evolutionary consequences of ancient endogenous retroviruses. Nat. Rev. Microbiol. 2019, 17, 355–370. [Google Scholar] [CrossRef]
- Yang, L.; Malhotra, R.; Chikhi, R.; Elleder, D.; Kaiser, T.; Rong, J.; Medvedev, P.; Poss, M. Recombination marks the evolutionary dynamics of a recently endogenized retrovirus. Mol. Biol. Evol. 2021, 38, 5423–5436. [Google Scholar] [CrossRef]
- Belshaw, R.; Katzourakis, A.; Paces, J.; Burt, A.; Tristem, M. High copy number in human endogenous retrovirus families is associated with copying mechanisms in addition to reinfection. Mol. Biol. Evol. 2005, 22, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Mayer, J.; Meese, E.U. The human endogenous retrovirus family HERV-K(HML-3). Genomics 2002, 80, 331–343. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| OWM Species | Specimen, Gender, Origin | No. of Full-Length, HOMOZYGOUS, Cer-SERV GENOMES 1 |
|---|---|---|
| Chlorocebus aethiops (vervet) | (a) #1994-021, male, Caribbean C. aethiops sabeus (b) WHO Vero cell line RCB 10–87, female sabaeus monkey 2 | (a) 6 (3 SERV-1, 3 SERV-2) (b) 6 (1 SERV-1, 5 SERV-2) |
| Erythrocebus patas (red guenon) | #BS28, unknown sex, San Diego Zoo | 1 (SERV-2) |
| Macaca fascicularis (crab-eating macaque) | #MacFas5, female, from Tinjil, Java, Indonesia | 2 (1 SERV-1, 1 SERV-2) |
| Macaca fuscata (Japanese macaque) | #JAMA01, unknown sex, Japan | 7 (4 SERV-1, 3 SERV-2) |
| Macaca mulatta (rhesus monkey) | #17573, female, Indian origin + separate Y chromosome | 9 (6 SERV-1, 3 SERV-2) |
| Macaca nemestrina (pig-tailed macaque) | #M95218, female, Washington National Primate Research Center | 1 (SERV-1) 3 |
| Mandrillus leucophaeus (drill) | Isolate #KB7577, female, San Diego Zoo | 1 (SERV-2) 3 |
| Papio anubis (olive baboon) | (a) #X1155, female, Kenyan ancestry (b) #15944, male, Southwest National Primate Research Center | (a) 7 (5 SERV-1, 2 SERV-2) (b) 15 (12 SERV-1, 3 SERV-2) |
| Theropithecus gelada (gelada) | #Dixy, female, Ethiopia | 26 (21 SERV-1, 5 SERV-2) |
| Genotype/ No. of Sequences | Mean Group LTR nt Distance K 1 | LTR nt Distance K, Range | Calculated Integration Time (Fast Rate) 2 | Calculated Integration Time (Slow Rate) 2 |
|---|---|---|---|---|
| Cer-SERV-1 N = 55 | 0.023 ± 0.014 | 0.000–0.053 | 2.3 ± 1.4 mya (range <0.3–5.3) | 5.0 ± 3.0 mya (range <0.3–11.5) |
| Cer-SERV-2 N = 26 | 0.039 ± 0.019 | 0.011–0.084 | 3.9 ± 1.9 mya (range 2.4–18.0) | 8.5 ± 4.0 mya (range 2.4–18.0) |
| Chromosome 17 Cer-SERV-2 LTR | K (nt Distance) 1 | Estimated Age T of Integration 2 | Event, with Average Estimated Age |
|---|---|---|---|
| 5′/3′ LTR gelada | 0.072 | ~7.2/~15.7 mya | Integration of chr. 17 Cer-SERV-2 provirus: ~7.7/~16.8 mya |
| 5′/3′ LTR baboon 1 5′/3′ LTR baboon 2 | 0.084 0.075 | ~8.4/~18.3 mya ~7.5/~16.3 mya | |
| 5′ LTR gelada/ 5′ LTR baboon 1 5′ LTR gelada/ 5′ LTR baboon 2 | 0.037 0.028 | ~3.7/~8.0 mya ~2.8/~6.1 mya | TMRCA 3 of gelada and baboon chr. 17 Cer-SERV-2: ~2.3/~5.6 mya |
| 3′ LTR gelada/ 3′ LTR baboon 1 3′ LTR gelada/ 3′ LTR baboon 2 | 0.013 0.013 | ~1.3/~2.8 mya | |
| 5′ LTR baboon 1/ 5′ LTR baboon 2 | 0.019 | ~1.9/~4.1 mya | TMRCA of two P. anubis chr. 17 Cer-SERV-2: <0.8/~2.2 mya |
| 3′ LTR baboon 1/ 3′ LTR baboon 2 | 0.000 | <0.3 mya | |
| 5′ LTR gelada/ 5′ LTR mangabey | 0.047 | ~4.7/~10.2 mya | TMRCA gelada/baboon/mangabey chr. 17 Cer-SERV-2: ~3.8/~8.3 mya |
| 5′ LTR baboon 1/ 5′ LTR mangabey | 0.035 | ~3.5/~7.6 mya | |
| 5′ LTR baboon 2/ 5′ LTR mangabey | 0.033 | ~3.3/~7.2 mya |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Kuyl, A.C. Analysis of Simian Endogenous Retrovirus (SERV) Full-Length Proviruses in Old World Monkey Genomes. Genes 2022, 13, 119. https://doi.org/10.3390/genes13010119
van der Kuyl AC. Analysis of Simian Endogenous Retrovirus (SERV) Full-Length Proviruses in Old World Monkey Genomes. Genes. 2022; 13(1):119. https://doi.org/10.3390/genes13010119
Chicago/Turabian Stylevan der Kuyl, Antoinette C. 2022. "Analysis of Simian Endogenous Retrovirus (SERV) Full-Length Proviruses in Old World Monkey Genomes" Genes 13, no. 1: 119. https://doi.org/10.3390/genes13010119
APA Stylevan der Kuyl, A. C. (2022). Analysis of Simian Endogenous Retrovirus (SERV) Full-Length Proviruses in Old World Monkey Genomes. Genes, 13(1), 119. https://doi.org/10.3390/genes13010119