
Genome Chaos, Information Creation, and Cancer Emergence: Searching for New Frameworks on the 50th Anniversary of the “War on Cancer”



Abstract
:1. Introduction
“Looking back, it’s clear that the scramble to find human retroviruses represented the major irony of the War on Cancer: it had been launched for the wrong reason, since cancer-causing human retroviruses were never found (with the exception of rare leukemias in the Caribbean and southern Japan).”[2]
2. Newly Emergent Genomic and Evolutionary Concepts
2.1. Genes vs. Karyotypes (Chromosome Sets): Redefining Inheritance
2.2. Molecular vs. Evolutionary Mechanisms: The Parts/Pathways, the System, and the Selective Processes
2.3. Genome Chaos: The Essential Process of System Information Self-Creation
“Chaos as a system behavior does not simply mean random disorder. Why use the term “chaos” to describe genome-based rapid cancer evolution? Genome behavior during crises shares common features with complex systems described by chaos theory. Chaos theory, a non-linear dynamics concept, states that “within the apparent randomness of chaotic complex systems, there are underlying patterns, interconnectedness, constant feedback loops, repetition, self-similarity, fractals, and self-organization” (https://en.wikipedia.org/wiki/Chaos_theory) (accessed on 12 August 2021). It is important to note that, within the collective frameworks of chaos theory, apparently random states of disorder and irregularities of dynamic systems are often governed by deterministic laws. In layman’s terms, unfortunately, the word “chaos” suggests rampant disorder and randomness, coupled with a negative connotation”“…Genome chaos represents a process that can be described by a combination of uncertainty and certainty, disorder and order, stochasticity and determinism—an excellent example of random means to a nonrandom end function. Therefore, analyzing system behavior (how macroevolution is achieved by the success of creating new systems), rather than characterizing all involved genome variation mechanisms, will make the cellular macroevolution phase transition much easier to understand and predict.” [119];
2.4. Two-Phased Evolutionary Model
3. Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- National Cancer Act of 1971. Available online: https://www.cancer.gov/about-nci/overview/history/national-cancer-act-1971 (accessed on 12 August 2021).
- Weinberg, R.A. Coming Full Circle—From Endless Complexity to Simplicity and Back Again. Cell 2014, 157, 267–271. [Google Scholar] [CrossRef] [Green Version]
- Heng, H.H. Debating Cancer: The Paradox in Cancer Research; World Scientific Publishing Co.: Singapore, 2015; ISBN 978-981-4520-84-3. [Google Scholar]
- Ye, C.J.; Sharpe, Z.; Heng, H.H. Origins and Consequences of Chromosomal Instability: From Cellular Adaptation to Genome Chaos-Mediated System Survival. Genes 2020, 11, 1162. [Google Scholar] [CrossRef]
- Heng, H.H. Cancer genome sequencing: The challenges ahead. Bioessays 2007, 29, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H.; Stevens, J.B.; Liu, G.; Bremer, S.W.; Ye, K.J.; Reddy, P.-V.; Wu, G.S.; Wang, Y.A.; Tainsky, M.A.; Ye, C.J. Stochastic cancer progression driven by non-clonal chromosome aberrations. J. Cell. Physiol. 2006, 208, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H. The genome-centric concept: Resynthesis of evolutionary theory. Bioessays 2009, 31, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H. Genome Chaos: Rethinking Genetics, Evolution, and Molecular Medicine; Academic Press Elsevier: Cambridge, MA, USA, 2019; ISBN 978-012-8136-35-5. [Google Scholar]
- Mitelman, F. 50,000 Tumors, 40,000 Aberrations, and 300 Fusion Genes: How Much Remains? 50 Years of 46 Human Chromosomes: Progress in Cytogenetics; NCI, NIH: Bethesda, MD, USA, 2006. [Google Scholar]
- Park, S.Y.; Gönen, M.; Kim, H.J.; Michor, F.; Polyak, K. Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. J. Clin. Investig. 2010, 120, 636–644. [Google Scholar] [CrossRef] [Green Version]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [Green Version]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with re-duced response to immunotherapy. Science 2017, 355, eaaf8399. [Google Scholar] [CrossRef] [Green Version]
- Vargas-Rondón, N.; Villegas, V.E.; Rondón-Lagos, M. The role of chromosomalinstability in cancer and therapeutic responses. Cancers 2017, 10, 4. [Google Scholar] [CrossRef] [Green Version]
- Frias, S.; Ramos, S.; Salas, C.; Molina, B.; Sánchez, S.; Rivera-Luna, R. Nonclonal Chromosome Aberrations and Genome Chaos in Somatic and Germ Cells from Patients and Survivors of Hodgkin Lymphoma. Genes 2019, 10, 37. [Google Scholar] [CrossRef] [Green Version]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef]
- Yizhak, K.; Aguet, F.; Kim, J.; Hess, J.M.; Kübler, K.; Grimsby, J.; Frazer, R.; Zhang, H.; Haradhvala, N.J.; Rosebrock, D.; et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 2019, 364, eaaw0726. [Google Scholar] [CrossRef]
- de Magalhães, J.P. Every gene can (and possibly will) be associated with cancer. Trends Genet. 2021. [Google Scholar] [CrossRef]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef] [Green Version]
- McClellan, J.; King, M.-C. Genetic Heterogeneity in Human Disease. Cell 2010, 141, 210–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rancati, G.; Pavelka, N.; Fleharty, B.; Noll, A.; Trimble, R.; Walton, K.; Perera, A.; Staehling-Hampton, K.; Seidel, C.W.; Li, R. Aneuploidy Underlies Rapid Adaptive Evolution of Yeast Cells Deprived of a Conserved Cytokinesis Motor. Cell 2008, 135, 879–893. [Google Scholar] [CrossRef] [Green Version]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Chromosomal mosaicism goes global. Mol. Cytogenet. 2008, 1, 26. [Google Scholar] [CrossRef] [Green Version]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B.; Kutsev, S.I. Ontogenetic and Pathogenetic Views on Somatic Chromosomal Mosaicism. Genes 2019, 10, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, C.J.; Chen, J.; Liu, G.; Heng, H.H. Somatic Genomic Mosaicism in Multiple Myeloma. Front. Genet. 2020, 11, 388. [Google Scholar] [CrossRef] [Green Version]
- Nurse, P. Biology must generate ideas as well as data. Nature 2021, 597, 305. [Google Scholar] [CrossRef] [PubMed]
- Duesberg, P.; Rausch, C.; Rasnick, D.; Hehlmann, R. Genetic instability of cancer cells is proportional to their degree of aneuploidy. Proc. Natl. Acad. Sci. USA 1998, 95, 13692–13697. [Google Scholar] [CrossRef] [Green Version]
- Duesberg, P.; Rasnick, D. Aneuploidy, the somatic mutation that makes cancer a species of its own. Cell Motil Cytoskelet. 2000, 47, 81–107. [Google Scholar] [CrossRef]
- Gibbs, W.W. Untangling the roots of cancer. Sci. Am. 2003, 289, 56–65. [Google Scholar] [CrossRef]
- Weaver, B.A.; Cleveland, D.W. Aneuploidy: Instigator and Inhibitor of Tumorigenesis. Cancer Res. 2007, 67, 10103–10105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavelka, N.; Rancati, G.; Li, R. Dr Jekyll and Mr Hyde: Role of aneuploidy in cellular adaptation and cancer. Curr. Opin. Cell Biol. 2010, 22, 809–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, J.J.; Amon, A. New Insights into the Troubles of Aneuploidy. Annu. Rev. Cell Dev. Biol. 2012, 28, 189–214. [Google Scholar] [CrossRef] [Green Version]
- Ye, C.J.; Regan, S.; Liu, G.; Alemara, S.; Heng, H.H. Understanding aneuploidy in cancer through the lens of system inheritance, fuzzy inheritance and emergence of new genome systems. Mol. Cytogenet. 2018, 11, 31. [Google Scholar] [CrossRef]
- Soto, A.M.; Sonnenschein, C. The tissue organization field theory of cancer: A testable replacement for the somatic mutation theory. Bioessays 2011, 33, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Baker, S.G. TOFT better explains experimental results in cancer research than SMT. Bioessays 2011, 33, 919–921. [Google Scholar] [CrossRef]
- Soto, A.M.; Sonnenschein, C. Paradoxes in Carcinogenesis: There Is Light at the End of That Tunnel! Disruptive Sci. Technol. 2013, 1, 154–156. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Ernberg, I.; Kauffman, S. Cancer attractors: A systems view of tumors from a gene network dynamics and developmental perspective. Semin. Cell Dev. Biol. 2009, 20, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Huang, S. Genetic and non-genetic instability in tumor progression: Link between the fitness landscape and the epigenetic landscape of cancer cells. Cancer Metastasis Rev. 2013, 32, 423–448. [Google Scholar] [CrossRef]
- Kulkarni, P.; Shiraishi, T.; Kulkarni, R.V. Cancer: Tilting at windmills? Mol. Cancer 2013, 12, 108. [Google Scholar] [CrossRef] [Green Version]
- Ao, P.; Galas, D.; Hood, L.; Yin, L.; Zhu, X.M. Towards predictive stochastic dynamical modeling of cancer genesis and progression. Interdiscip. Sci. Comput. Life Sci. 2010, 2, 140–144. [Google Scholar] [CrossRef] [Green Version]
- Noble, D. Cellular Darwinism: Regulatory networks, stochasticity, and selection in cancer development. Prog. Biophys. Mol. Biol. 2021, 165, 66–71. [Google Scholar] [CrossRef]
- Davies, P.C.; Lineweaver, C.H. Cancer tumors as Metazoa 1.0: Tapping genes of ancient ancestors. Phys. Biol. 2011, 8, 015001. [Google Scholar] [CrossRef]
- Wilkins, A.S. The enemy within: An epigenetic role of retrotransposons in cancer initiation. Bioessays 2010, 32, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Beedle, A.S.; Hanson, M.A.; Low, F.M. Human Growth: Evolutionary and Life History Perspectives. Nestle Nutr. Inst. Workshop Ser. 2013, 71, 89–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horne, S.D.; Chowdhury, S.K.; Heng, H.H. Stress, genomic adaptation, and the evolutionary trade-off. Front Genet. 2014, 5, 92. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, L.F. A Calcium-Based Theory of Carcinogenesis. Adv. Cancer Res. 2005, 94, 231–263. [Google Scholar] [CrossRef]
- Levin, M. Bioelectrical approaches to cancer as a problem of the scaling of the cellular self. Prog. Biophys. Mol. Biol. 2021, 165, 102–113. [Google Scholar] [CrossRef]
- Ewald, P.W. An evolutionary perspective on parasitism as a cause of cancer. Adv. Parasitol. 2009, 68, 21–43. [Google Scholar] [CrossRef]
- Warburg, O. On Respiratory Impairment in Cancer Cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef] [PubMed]
- A Loeb, L.; Springgate, C.F.; Battula, N. Errors in DNA replication as a basis of malignant changes. Cancer Res. 1974, 34, 2311–2321. [Google Scholar]
- Heppner, G.H. Tumor heterogeneity. Cancer Res. 1984, 44, 2259–2265. [Google Scholar] [PubMed]
- Heppner, G.H.; Miller, B.E. Therapeutic implications of tumor heterogeneity. Semin. Oncol. 1989, 16, 91–105. [Google Scholar] [PubMed]
- Raza, A. The First Cell: And the Human Costs of Pursuing Cancer to the Last. Basic Books; Hachette Book Group, Inc.: New York, NY, USA, 2019. [Google Scholar]
- Heng, J.; Heng, H.H. Two-phased evolution: Genome chaos-mediated information creation and maintenance. Prog. Biophys. Mol. Biol. 2021, 165, 29–42. [Google Scholar] [CrossRef]
- Heng, H.H.; Liu, G.; Stevens, J.B.; Abdallah, B.Y.; Horne, S.D.; Ye, K.J.; Bremer, S.W.; Chowdhury, S.K.; Ye, C.J. Karyotype heterogeneity and unclassified chromosomal abnormalities. Cytogenet. Genome Res. 2013, 139, 144–157. [Google Scholar] [CrossRef]
- Heng, H.; Chen, W.Y.; Wang, Y.C. Effects of pingyanymycin on chromosomes: A possible structural basis for chromosome aberration. Mutat. Res. Mol. Mech. Mutagen. 1988, 199, 199–205. [Google Scholar] [CrossRef]
- Heng, H.H. Chapter 12: Bio-complexity challenging reductionism. In Handbook on System and Complexity in Health; Sturmberg, J., Martin, C., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 193–208. [Google Scholar]
- Tez, M.; Tez, S. Cancer is the chaotic search for adaptation to previously unknown environments. Theor. Biol. Forum 2016, 109, 149–154. [Google Scholar]
- Wu, S.; Turner, K.M.; Nguyen, N.-P.; Raviram, R.; Erb, M.; Santini, J.; Luebeck, J.; Rajkumar, U.; Diao, Y.; Li, B.; et al. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 2019, 575, 699–703. [Google Scholar] [CrossRef]
- Raghuram, G.V.; Chaudhary, S.; Johari, S.; Mittra, I. Illegitimate and Repeated Genomic Integration of Cell-Free Chromatin in the Aetiology of Somatic Mosaicism, Ageing, Chronic Diseases and Cancer. Genes 2019, 10, 407. [Google Scholar] [CrossRef] [Green Version]
- Huxley, J. Cancer Biology: Comparative and Genetic. Biol. Rev. 1956, 31, 474–514. [Google Scholar] [CrossRef]
- van Valen, L.; Mairorana, V.C. Hela, a new microbial species. Evol Theor. 1991, 10, 71–74. [Google Scholar]
- Ye, C.J.; Liu, G.; Bremer, S.W.; Heng, H.H.Q. The dynamics of cancer chromosomes and genomes. Cytogenet. Genome Res. 2007, 118, 237–246. [Google Scholar] [CrossRef]
- Heng, H.H. Elimination of altered karyotypes by sexual reproduction preserves species identity. Genome 2007, 50, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.D. The Animal within: Carcinogenesis and the Clonal Evolution of Cancer Cells Are Speciation Events Sensu Stricto. Evolution 2010, 64, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Bloomfield, M.; Duesberg, P. Inherent variability of cancer-specific aneuploidy generates metastases. Mol. Cytogenet. 2016, 9, 90. [Google Scholar] [CrossRef] [Green Version]
- Paul, D. Cancer as a form of life: Musings of the cancer and evolution symposium. Prog. Biophys. Mol. Biol. 2021, 165, 120–139. [Google Scholar] [CrossRef]
- Walen, K.H. Mitosis is not the only distributor of mutated cells: Non-mitotic endopolyploid cells produce reproductive genome-reduced cells. Cell Biol. Int. 2010, 34, 867–872. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Kalejs, M.; Cragg, M. Mitotic catastrophe and endomitosis in tumour cells: An evolutionary key to a molecular solution. Cell Biol. Int. 2005, 29, 1012–1018. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Salmina, K.; Anatskaya, O.; Cragg, M.S. Paradoxes of cancer: Survival at the brink. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Heng, H.H.; Stevens, J.B.; Lawrenson, L.; Liu, G.; Ye, K.J.; Bremer, S.W.; Ye, C.J. Patterns of genome dynamics and cancer evolution. Cell. Oncol. Off. J. Int. Soc. Cell. Oncol. 2008, 30, 513–514. [Google Scholar] [CrossRef]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene 2013, 33, 116–128. [Google Scholar] [CrossRef]
- Niu, N.; Mercado-Uribe, I.; Liu, J. Dedifferentiation into blastomere-like cancer stem cells via formation of polyploid giant cancer cells. Oncogene 2017, 36, 4887–4900. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Niu, N.; Zhang, J.; Qi, L.; Shen, W.; Donkena, K.V.; Feng, Z.; Liu, J. Polyploid Giant Cancer Cells (PGCCs): The Evil Roots of Cancer. Curr. Cancer Drug Targets 2019, 19, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Liu, J. The dualistic origin of human tumors. Semin. Cancer Biol. 2018, 53, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, J. The “life code”: A theory that unifies the human life cycle and the origin of human tumors. Semin. Cancer Biol. 2020, 60, 380–397. [Google Scholar] [CrossRef]
- Ye, J.C.; Horne, S.; Zhang, J.Z.; Jackson, L.; Heng, H.H. Therapy Induced Genome Chaos: A Novel Mechanism of Rapid Cancer Drug Resistance. Front. Cell Dev. Biol. 2021, 9, 676344. [Google Scholar] [CrossRef]
- Zaitceva, V.; Kopeina, G.S.; Zhivotovsky, B. Anastasis: Return Journey from Cell Death. Cancers 2021, 13, 3671. [Google Scholar] [CrossRef]
- Stevens, J.B.; Liu, G.; Bremer, S.W.; Ye, K.J.; Xu, W.; Xu, J.; Sun, Y.; Wu, G.S.; Savasan, S.; Krawetz, S.A.; et al. Mitotic Cell Death by Chromosome Fragmentation. Cancer Res. 2007, 67, 7686–7694. [Google Scholar] [CrossRef] [Green Version]
- Pienta, K.; Hammarlund, E.; Austin, R.; Axelrod, R.; Brown, J.; Amend, S. Cancer cells employ an evolutionarily conserved polyploidization program to resist therapy. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [Green Version]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, J.A. What can evolutionary biology learn from cancer biology? Prog. Biophys. Mol. Biol. 2021, 165, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, J.; Noble, D. The value of treating cancer as an evolutionary disease. Prog. Biophys. Mol. Biol. 2021, 165, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Furst, R. The Importance of Henry H. Heng’s Genome Architecture Theory. Prog. Biophys. Mol. Biol. 2021, 165, 153–156. [Google Scholar] [CrossRef]
- Vendramin, R.; Litchfield, K.; Swanton, C. Cancer evolution: Darwin and beyond. EMBO J. 2021, 40, e108389. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.J.; Stilgenbauer, L.; Moy, A.; Liu, G.; Heng, H.H. What Is Karyotype Coding and Why Is Genomic Topology Important for Cancer and Evolution? Front. Genet. 2019, 10, 1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boveri, T. Concerning the Origin of Malignant Tumours by Theodor Boveri. Translated and annotated by Henry Harris. J. Cell Sci. 2008, 121 (Suppl. 1), 1–84. [Google Scholar] [CrossRef]
- Heng, H.H.; Liu, G.; Stevens, J.B.; Bremer, S.W.; Ye, K.J.; Abdallah, B.Y.; Horne, S.D.; Ye, C.J. Decoding the genome beyond sequencing: The new phase of genomic research. Genomics 2011, 98, 242–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, C.J.; Stevens, J.B.; Liu, G.; Bremer, S.W.; Jaiswal, A.S.; Ye, K.J.; Lin, M.-F.; Lawrenson, L.; Lancaster, W.D.; Kurkinen, M.; et al. Genome based cell population heterogeneity promotes tumorigenicity: The evolutionary mechanism of cancer. J. Cell. Physiol. 2008, 219, 288–300. [Google Scholar] [CrossRef] [Green Version]
- Heng, H.H.; Bremer, S.W.; Stevens, J.B.; Horne, S.D.; Liu, G.; Abdallah, B.Y.; Ye, K.J.; Ye, C.J. Chromosomal instability (CIN): What it is and why it is crucial to cancer evolution. Cancer Metastasis Rev. 2013, 32, 325–340. [Google Scholar] [CrossRef]
- Liu, G.; Stevens, J.B.; Horne, S.D.; Abdallah, B.Y.; Ye, K.J.; Bremer, S.W.; Ye, C.J.; Chen, D.J.; Heng, H.H. Genome chaos: Survival strategy during crisis. Cell Cycle 2014, 13, 528–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, J. How Chaotic Is Genome Chaos? Cancers 2021, 13, 1358. [Google Scholar] [CrossRef] [PubMed]
- Pellestor, F.; Gatinois, V. Chromoanagenesis: A piece of the macroevolution scenario. Mol. Cytogenet. 2020, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Pellestor, F.; Gaillard, J.; Schneider, A.; Puechberty, J.; Gatinois, V. Chromoanagenesis, the mechanisms of a genomic chaos. Semin. Cell Dev. Biol. 2021. [Google Scholar] [CrossRef]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Horne, S.D.; Heng, H.H. Genome Chaos, Chromothripsis and Cancer Evolution. J. Cancer Stud. Ther. 2014, 1, 1–6. [Google Scholar]
- Heng, H.H. Chapter 5: Cancer Genomic Landscape, Ecology and Evolution of Cancer; Ujvari, B., Roche, B., Thomas, F., Eds.; Elseiver: Amsterdam, The Netherlands, 2017; pp. 69–86. [Google Scholar]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; Macdonald, T.Y.; Ghandi, M.; et al. Punctuated Evolution of Prostate Cancer Genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [Green Version]
- Crasta, K.; Ganem, N.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef]
- Forment, J.V.; Kaidi, A.; Jackson, S.P. Chromothripsis and cancer: Causes and consequences of chromosome shattering. Nat. Rev. Cancer 2012, 12, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.J.; Cleveland, D.W. Chromoanagenesis and cancer: Mechanisms and consequences of localized, complex chromosomal rearrangements. Nat. Med. 2012, 18, 1630–1638. [Google Scholar] [CrossRef] [Green Version]
- Inaki, K.; Liu, E.T. Structural mutations in cancer: Mechanistic and functional insights. Trends Genet. 2012, 28, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.J.; Jallepalli, P.V. Chromothripsis: Chromosomes in crisis. Dev. Cell. 2012, 23, 908–917. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Erez, A.; Nagamani, S.C.S.; Dhar, S.U.; Kołodziejska, K.E.; Dharmadhikari, A.V.; Cooper, M.L.; Wiszniewska, J.; Zhang, F.; Withers, M.A.; et al. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell 2001, 146, 889–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, A.; Lindberg, M.; Faust, G.G.; Leibowitz, M.L.; Clark, R.A.; Layer, R.M.; Quinlan, A.R.; Hall, I.M. Breakpoint profiling of 64 cancer genomes reveals numerous complex rear-rangements spawned by homology-independent mechanisms. Genome Res. 2013, 23, 762–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Righolt, C.; Mai, S. Shattered and stitched chromosomes-chromothripsis and chromoanasynthesis-manifestations of a new chromosome crisis? Genes Chromosomes Cancer 2012, 51, 975–981. [Google Scholar] [CrossRef] [PubMed]
- Setlur, S.R.; Lee, C. Tumor Archaeology Reveals that Mutations Love Company. Cell 2012, 149, 959–961. [Google Scholar] [CrossRef] [Green Version]
- Tubio, J.M.; Estivill, X. Cancer: When catastrophe strikes a cell. Nature 2011, 470, 476–477. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H. Karyotypic Chaos, A Form of Non-Clonal Chromosome Aberrations, Plays A Key Role for Cancer Progression and Drug Resistance; FASEB: Nuclear Structure and Cancer; Vermont Academy: Saxtons River, VT, USA, 2009. [Google Scholar]
- Heng, H.H.; Regan, S.M.; Liu, G.; Ye, C.J. Why it is crucial to analyze non clonal chromosome aberrations or NCCAs? Mol. Cytogenet. 2016, 9, 15. [Google Scholar] [CrossRef] [Green Version]
- Duesberg, P. Chromosomal Chaos and Cancer. Sci. Am. 2007, 296, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef] [Green Version]
- Heng, H.H.; Stevens, J.B.; Bremer, S.W.; Liu, G.; Abdallah, B.Y.; Ye, C.J. Evolutionary Mechanisms and Diversity in Cancer. Adv. Cancer Res. 2011, 112, 217–253. [Google Scholar] [CrossRef] [PubMed]
- Morishita, M.; Muramatsu, T.; Suto, Y.; Hirai, M.; Konishi, T.; Hayashi, S.; Shigemizu, D.; Tsunoda, T.; Moriyama, K.; Inazawa, J. Chromothripsis-like chromosomal rearrangements induced by ionizing radiation using proton microbeam irradiation system. Oncotarget 2016, 7, 10182–10192. [Google Scholar] [CrossRef]
- Mardin, B.R.; Drainas, A.P.; Waszak, S.M.; Weischenfeldt, J.; Isokane, M.; Stütz, A.M.; Raeder, B.; Efthymiopoulos, T.; Buccitelli, C.; Se-gura-Wang, M.; et al. A cell-based model system links chromothripsis with hyperploidy. Mol. Syst. Biol. 2015, 11, 828. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.J.; Liu, G.; Heng, H.H. Experimental Induction of Genome Chaos. Methods Mol. Biol. 2018, 337–352. [Google Scholar] [CrossRef]
- Ye, C.J.; Sharpe, Z.; Alemara, S.; Mackenzie, S.; Liu, G.; Abdallah, B.; Horne, S.; Regan, S.; Heng, H.H. Micronuclei and Genome Chaos: Changing the System Inheritance. Genes 2019, 10, 366. [Google Scholar] [CrossRef] [Green Version]
- Heng, J.; Heng, H.H. Genome chaos: Creating new genomic information essential for cancer macroevolution. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Heng, J.; Heng, H.H. Karyotype coding: The creation and maintenance of system information for complexity and biodiversity. Biosystems 2021, 208, 104476. [Google Scholar] [CrossRef]
- Barbieri, M. What is code biology? Biosystems 2018, 164, 1–10. [Google Scholar] [CrossRef]
- Barbieri, M. Overview of the third special issue in code biology. Biosystems 2021, 210, 104553. [Google Scholar] [CrossRef]
- Barbieri, M. A new theory of development: The generation of complexity in ontogenesis. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crkvenjakov, R.; Heng, H.H. Further illusions: On key evolutionary mechanisms that could never fit with Modern Synthesis. Prog. Biophys. Mol. Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, A.S.; Holliday, R. The Evolution of Meiosis from Mitosis. Genetics 2009, 181, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorelick, R.; Heng, H.H. Sex Reduces Genetic Variation: A Multidisciplinary Review. Evolution 2010, 65, 1088–1098. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Heng, E.; Moy, A.; Liu, G.; Heng, H.H.; Zhang, K. ER Stress and Micronuclei Cluster: Stress Response Contributes to Genome Chaos in Cancer. Front. Cell Dev. Biol. 2021, 9, 673188. [Google Scholar] [CrossRef]
- The White House Office of the Press Secretary, 2000. Available online: https://www.genome.gov/10001356/june-2000-white-house-event (accessed on 1 December 2021).
- Transcript: President Obama’s Final State of the Union Address. Available online: https://www.npr.org/2016/01/12/462831088/president-obama-state-of-the-union-transcript (accessed on 8 December 2021).
- Gatenby, R.A.; Silva, A.S.; Gillies, R.J.; Frieden, B.R. Adaptive therapy. Cancer Res. 2009, 69, 4894–4903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://www.cityofhope.org/breakthroughs/the-war-on-cancer-at-50-where-are-we (accessed on 12 August 2021).
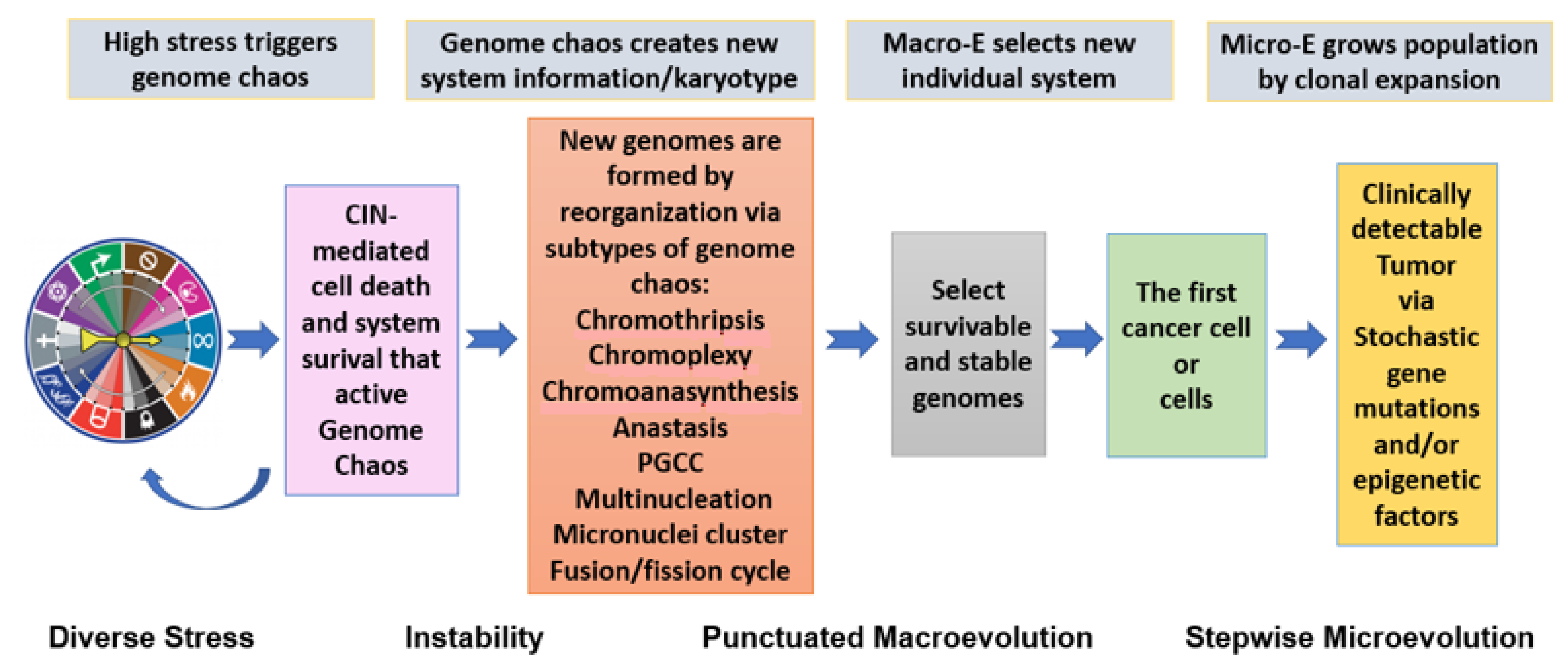

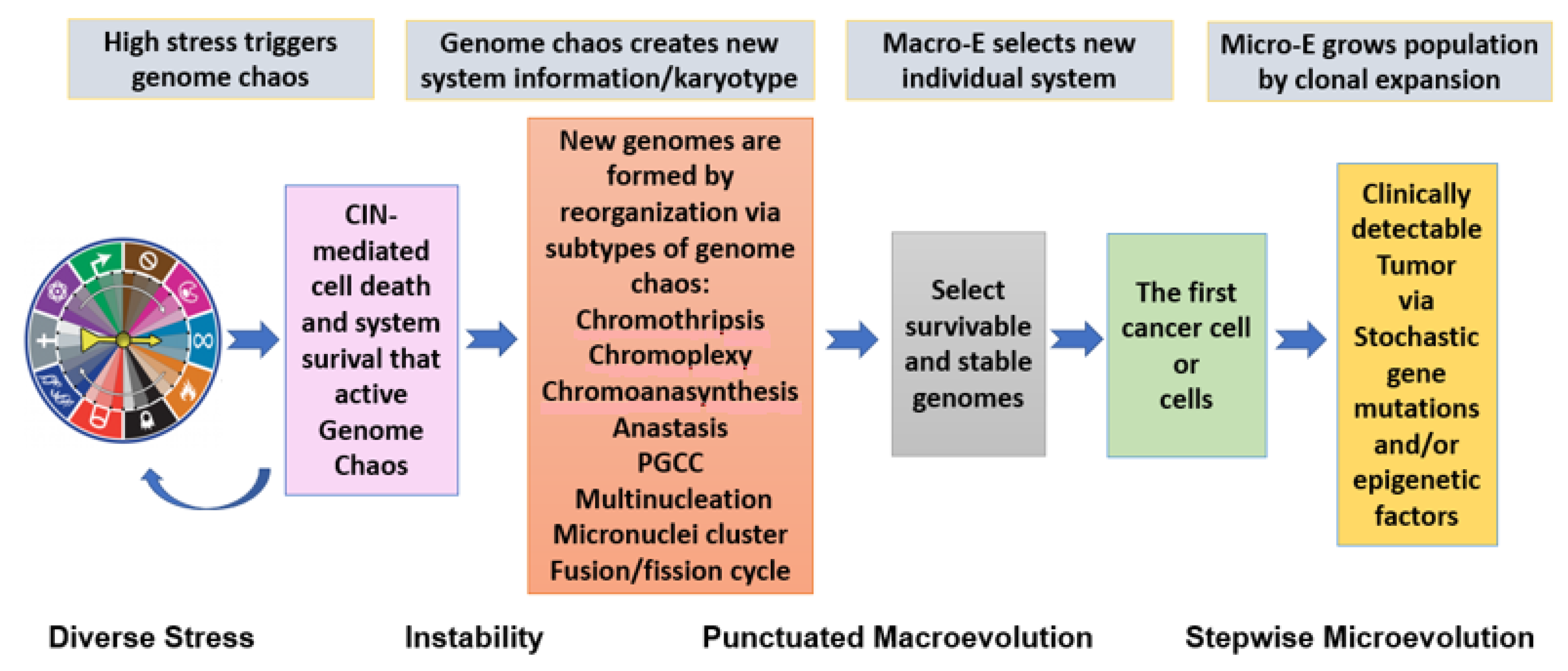{kind=link}
| Examples of Alternative Theories/Concepts for Explaining Cancer | Ref. | |
|---|---|---|
| Aneuploidy Theory | Duesberg et al. (1998) | [25] |
| Duesberg and Rasnick (2000) | [26] | |
| Gibbs (2003) | [27] | |
| Weaver and Cleveland (2007) | [28] | |
| Pavelka et al. (2010) | [29] | |
| Siegel and Amon (2012) | [30] | |
| Ye et al. (2018) | [31] | |
| Tissue organization field theory (TOFT) | Soto and Sonnenschein, (2011) | [32] |
| Baker (2011) | [33] | |
| Soto and Sonnenschein (2013) | [34] | |
| Cancer attractor theory | Huang et al. (2009) | [35] |
| Huang (2013) | [36] | |
| Kulkarni et al. (2013) | [37] | |
| Endogenous network hypothesis | Ao et al. (2010) | [38] |
| Physiological regulatory networks | Noble (2021) | [39] |
| Cancer represents a type of atavism | Davies (2021) | [40] |
| Retrotransposon-mediated genome evolution | Wilkins (2010) | [41] |
| Human genome mismatches the changing environment | Gluckman (2011) | [42] |
| Cancer represents a trade-off of adaptation and survival | Horne et al. (2014) | [43] |
| Heng (2015), (2019) | [3,8] | |
| Epigenetic alterations drive cancer | Feinberg et al. (2006) | [44] |
| Physical or chemical triggers | Jaffe (2005) | [45] |
| Levin (2021) | [46] | |
| Infection | Ewald (1998) | [47] |
| The Warburg effect and metabolic contribution | Warburg (1956) | [48] |
| Mutator phenotype | Loeb (1974) | [49] |
| Heterogeneity and tumor society, fuzzy inheritance | Heppner (1984), Heppner and Miller (1988) | [50,51] |
| Heng (2015) | [3] | |
| The first cell | Raza (2019) | [52] |
| New system emergent from system constraints | Heng and Heng (2021) | [53] |
| Complex adaptive system theory, chaotic systems | Heng (2006), (2013), (2019) Tez 2016 | [8,54,55] [56] [57] |
| Extrachromosomal DNA | Wu et al. (2019) | [58] |
| Illegitimate genomic integration of cell-free chromatin | Raghuram et al., (2019) | [59] |
| Cancer as a new cellular species | Huxley (1956) | [60] |
| Van Valen (1991) | [61] | |
| Duesberg and Rasnick (2000) | [26] | |
| Ye et al. (2007) | [62] | |
| Heng (2007) | [63] | |
| Vincent (2010) | [64] | |
| Heng (2015) | [3] | |
| Bloomfield and Duesberg (2016) | [65] | |
| Paul (2021) | [66] | |
| Chaotic genomes (structural and numerical subtypes): Massive karyotype reorganization; Polyploidy giant cancer cells; Micronuclei clusters; Chromosome fragmentations; Anstasis; and other overlooked chromosomal and nuclear variations | Walen (2010) | [67] |
| Erenpreisa et al. (2005) (2020) | [68,69] | |
| Heng et al. (1988), (2006), (2008), (2013) | [6,54,55,70] | |
| Zhang et al. (2014) | [71] | |
| Niu et al. (2016) | [72] | |
| Chen et al. (2019) | [73] | |
| Liu (2018) | [74] | |
| Liu (2020) | [75] | |
| Ye et al. (2019) | [76] | |
| Zaitceva et al. (2021) | [77] | |
| Stevens et al. (2007) | [78] | |
| Pienta et al. (2020) | [79] | |
| Punctuated macroevolution | Heng et al. (2006) | [6] |
| Navin et al. (2011) | [80] | |
| Sottoriva et al. (2015) | [81] | |
| Shapiro (2021) | [82] | |
| Shapiro and Noble (2021) | [83] | |
| Furst (2021) | [84] | |
| Vendramin et al. (2021) | [85] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heng, J.; Heng, H.H. Genome Chaos, Information Creation, and Cancer Emergence: Searching for New Frameworks on the 50th Anniversary of the “War on Cancer”. Genes 2022, 13, 101. https://doi.org/10.3390/genes13010101
Heng J, Heng HH. Genome Chaos, Information Creation, and Cancer Emergence: Searching for New Frameworks on the 50th Anniversary of the “War on Cancer”. Genes. 2022; 13(1):101. https://doi.org/10.3390/genes13010101
Chicago/Turabian StyleHeng, Julie, and Henry H. Heng. 2022. "Genome Chaos, Information Creation, and Cancer Emergence: Searching for New Frameworks on the 50th Anniversary of the “War on Cancer”" Genes 13, no. 1: 101. https://doi.org/10.3390/genes13010101
APA StyleHeng, J., & Heng, H. H. (2022). Genome Chaos, Information Creation, and Cancer Emergence: Searching for New Frameworks on the 50th Anniversary of the “War on Cancer”. Genes, 13(1), 101. https://doi.org/10.3390/genes13010101