
A Rare Case of Brachyolmia with Amelogenesis Imperfecta Caused by a New Pathogenic Splicing Variant in LTBP3

 ,
,  , ,
, ,  , and
, and
Abstract
:1. Introduction
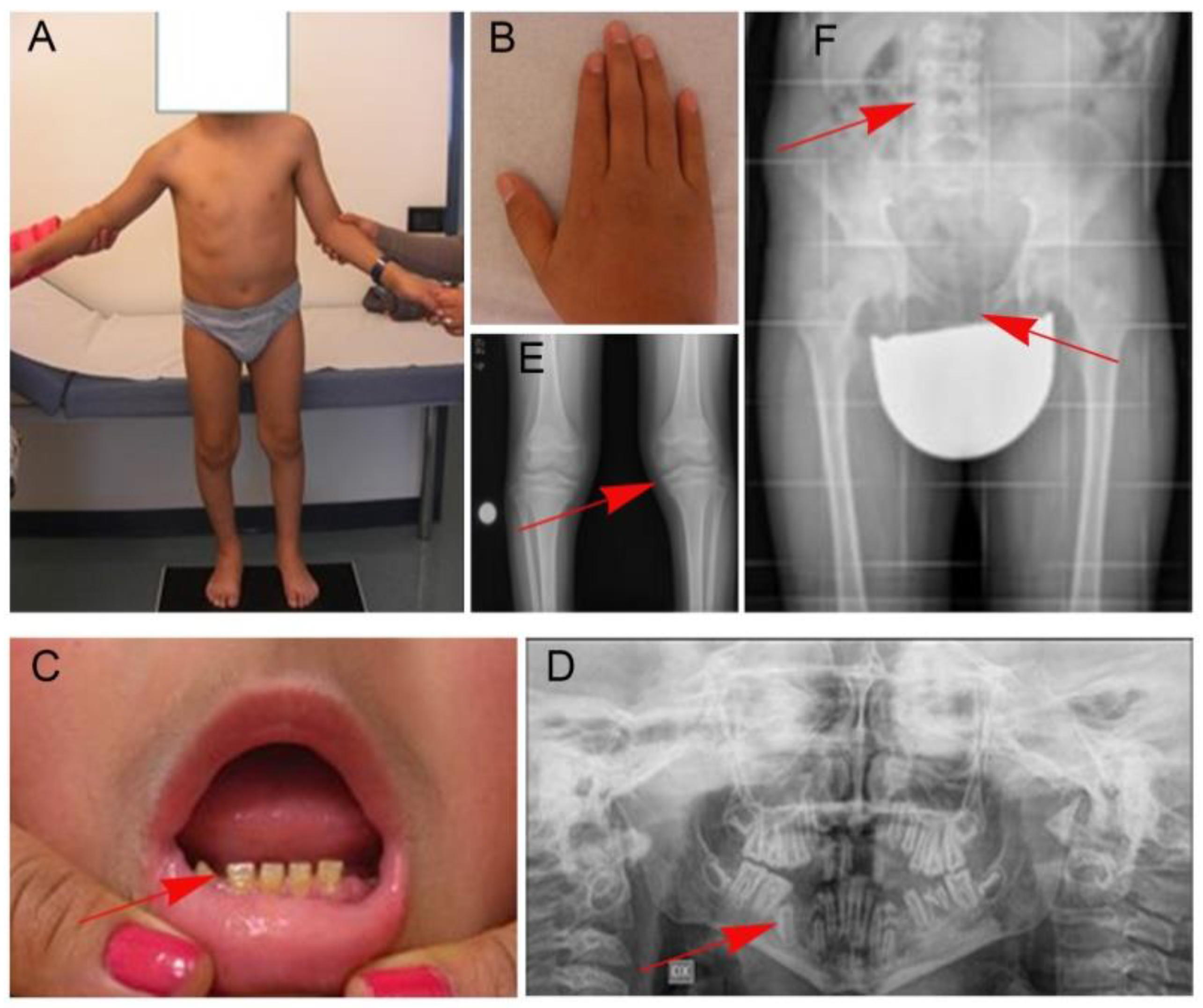
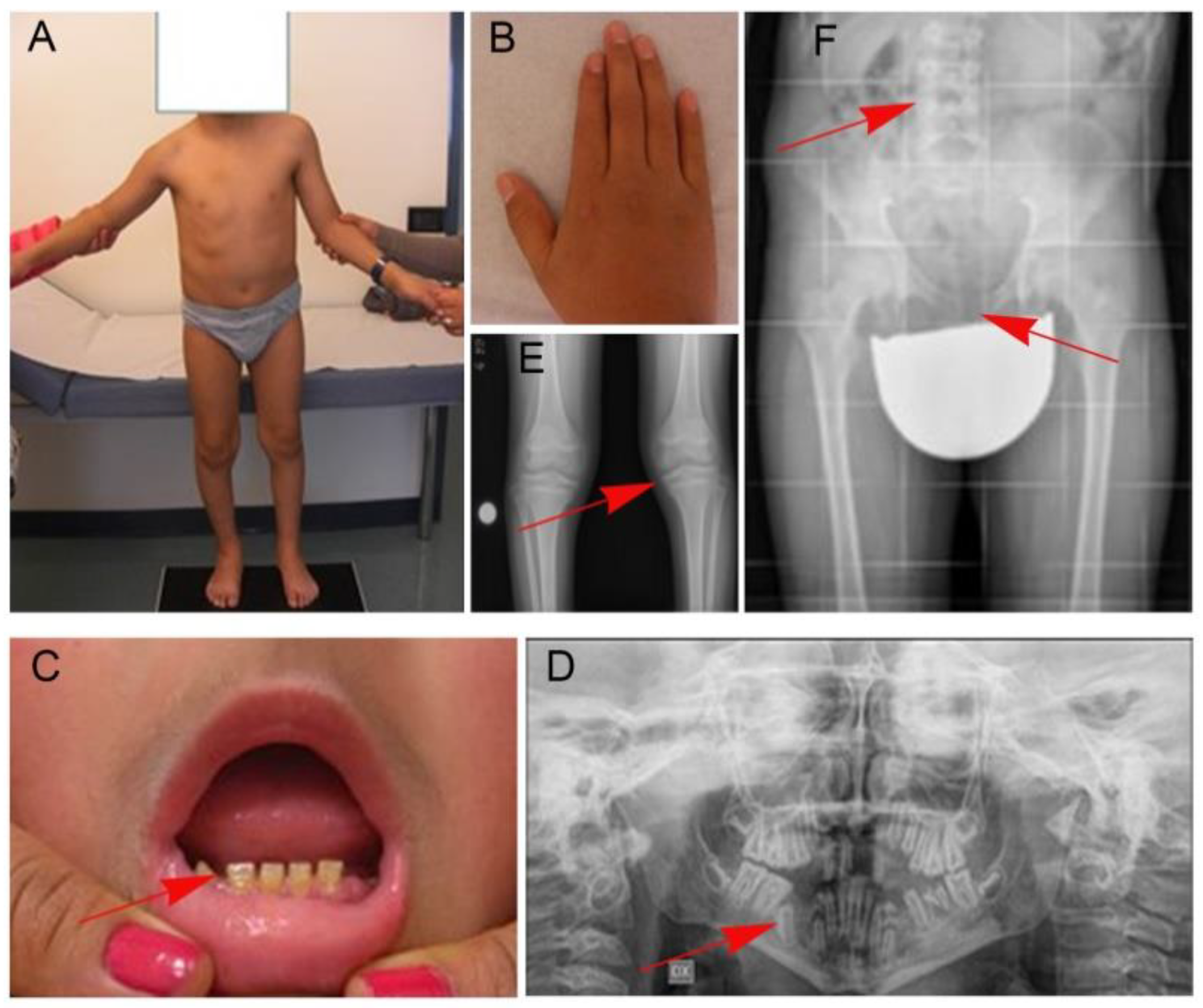
2. Patient
3. Materials and Methods
3.1. Samples and RNA/DNA Extraction
3.2. Comparative Genomic Hybridization Analysis
3.3. Whole Exome Sequencing
3.4. Sanger Sequencing
3.5. RNA Analysis
4. Results
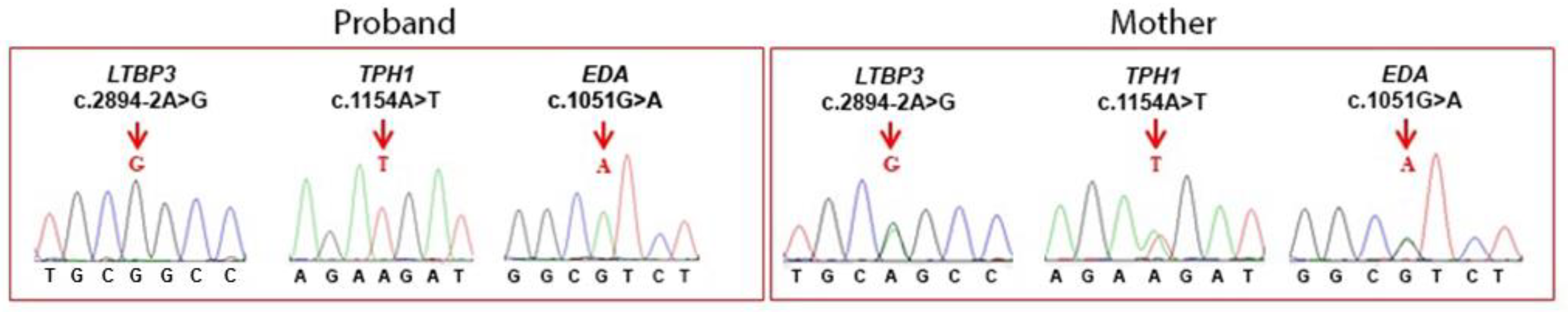
4.1. WES Analysis
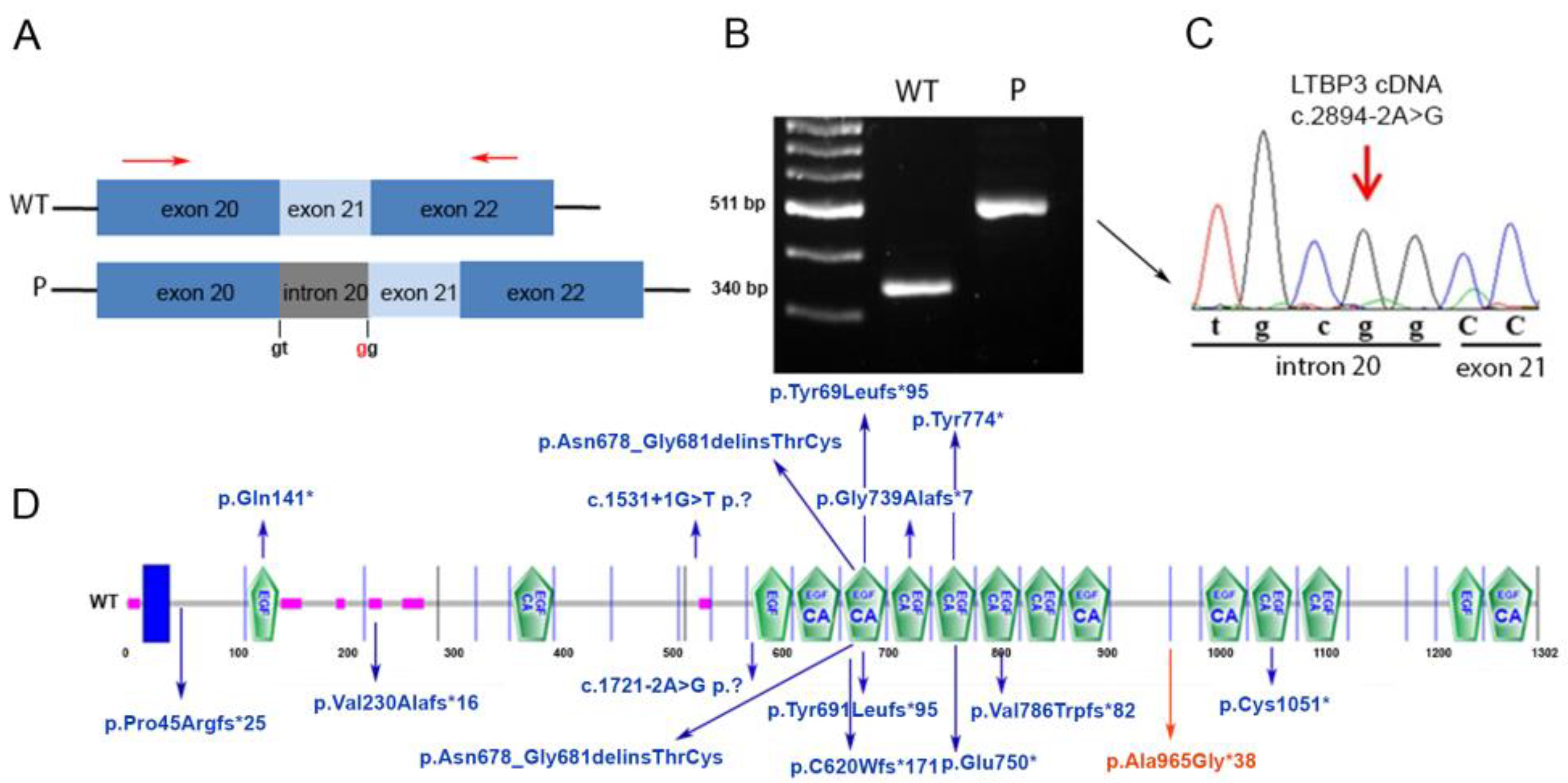
4.2. RNA Analysis
5. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shohat, M.; Lachman, R.; Gruber, H.E.; Rimoin, D.L. Brachyolmia: Radiographic and genetic evidence of heterogeneity. Am. J. Med. Genet. 1989, 33, 209–219. [Google Scholar] [CrossRef]
- Bownass, L.; Abbs, S.; Armstrong, R.; Baujat, G.; Behzadi, G.; Berentsen, R.D.; Burren, C.; Calder, A.; Cormier-Daire, V.; Newbury-Ecob, R.; et al. PAPSS2-related brachyolmia: Clinical and radiological phenotype in 18 new cases. Am. J. Med. Genet. 2019, 179, 1884–1894. [Google Scholar] [CrossRef]
- Rock, M.J.; Prenen, J.; Funari, V.A.; Funari, T.L.; Merriman, B.; Nelson, S.F.; Lachman, R.S.; Wilcox, W.R.; Reyno, S.; Quadrelli, R.; et al. Gain-of-function mutations in TRPV4 cause autosomal dominant brachyolmia. Nat. Genet. 2008, 40, 999–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfred, M.J.; Crawford, P.J.M.; Savarirayan, R. Amelogenesis imperfecta—A classification and catalogue for the 21st century. Oral Dis. 2003, 9, 19–23. [Google Scholar] [CrossRef]
- Crawford, P.J.M.; Aldred, M.; Bloch-Zupan, A. Amelogenesis imperfecta. Orphanet J. Rare Dis. 2007, 2, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, T.C.; Hart, P.S.; Gorry, M.C.; Michalec, M.D.; Ryu, O.H.; Uygur, C.; Ozdemir, D.; Firatli, S.; Aren, G.; Firatli, E. Novel ENAM mutation responsible for autosomal recessive amelogenesis imperfecta and localised enamel defects. J. Med. Genet. 2003, 40, 900–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verloes, A.; Jamblin, P.; Koulischer, L.; Bourguignon, J.P. A new form of skeletal dysplasia with amelogenesis imperfecta and platyspondyly. Clin. Genet. 1996, 49, 2–5. [Google Scholar] [CrossRef]
- Bertola, D.R.; Antequera, R.; Rodovalho, M.J.; Honjo, R.S.; Albano, L.M.J.; Furquim, I.M.; Oliveira, L.A.; Kim, C.A. Brachyolmia with amelogenesis imperfecta: Further evidence of a distinct entity. Am. J. Med. Genet. 2009, 149A, 532–534. [Google Scholar] [CrossRef]
- Noor, A.; Windpassing, C.; Victu, I.; Orlic, M.; Arshad Rafiq, M.; Khalid, M.; Nasir, M.N.; Ayub, M.; Alman, B.; Vicent, J.B. Oligodontiais caused by mutation in LTBP3, the gene encoding latetnt TGF-β binding protein 3. Am. J. Hum. Genet. 2009, 84, 519–523. [Google Scholar] [CrossRef] [Green Version]
- Huckert, M.; Stoetzel, C.; Morkumued, S.; Luagel-Haushalter, V.; Geoffroy, V.; Muller, J.; Clauss, F.; Prasad, M.K.; Obry, F.; Raymond, J.L.; et al. Mutation in the latent TGF-β binding protein 3(LTBP3) genecausebrachyolmia with amelogenesis imperfecta. Hum. Mol. Genet. 2015, 24, 3038–3049. [Google Scholar] [CrossRef] [Green Version]
- Dugan, S.L.; Temme, R.T.; Olson, R.A.; Mikhailov, A.; Law, R.; Mahmood, H.; Noor, A.; Vincent, J.B. New recessive truncating mutation in LTBP3 in a family with oligodontia, short stature, and mitral valve prolapse. Am. J. Med. Genet. 2015, 167A, 1396–1399. [Google Scholar] [CrossRef]
- Guo, D.C.; Regalado, E.S.; Pinard, A.; Chen, J.; Lee, K.; Rigelsky, C.; Zilbeberg, L.; Hostetler, E.M.; Aldred, M.; Wallace, S.E.; et al. LTBP3 pathogenic variants predispose individuals to thoracic aortic aneurysms and dissections. Am. J. Hum. Genet. 2018, 102, 706–712. [Google Scholar] [CrossRef] [Green Version]
- Faivre, L.; Le Merrer, M.; Baumann, C.; Polak, M.; Chatelain, P.; Sulmont, V.; Cousin, J.; Bost, M.; Cordier, M.; Zackai, E.; et al. Acromic dysplasia: Long term outcome and evidence of autosomal dominant inheritance. J. Med. Genet. 2001, 38, 745–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Goff, C.; Mahaut, C.; Wang, L.W.; Allali, S.; Abhyankar, A.; Jensen, S.; Zylberberg, L.; Collod-Beroud, G.; Bonnet, D.; Alanay, S.; et al. Mutations in the TGFbeta binding-protein-like domain 5 of FBN1 are responsible for acromicric and geleophysic dysplasias. Am. J. Hum. Genet. 2011, 89, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Intarak, N.; Theerapanon, T.; Thaweesapphithak, S.; Suphapeetiporn, K.; Porntaveetus, K.; Shotelersuk, V. Genotype-phenotype correlation and expansion of orodental anomalies in LTBP3-related disorders. Mol. Genet. Genom. 2019, 294, 773–787. [Google Scholar] [CrossRef]
- McInerney-Leo, A.M.; Le Goff, C.; Leo, P.L.; Kenna, T.J.; Keith, P.; Harris, J.E.; Steer, R.; Bole-Feysot, C.; Nitschke, P.; Kielty, C.; et al. Mutations in LTBP3 cause acromicric dysplasia and geleophysic dysplasia. J. Med. Gene. 2016, 53, 457–464. [Google Scholar] [CrossRef]
- Kaur, R.; Siddiqui, I.; Mathur, V.; Jana, M.; Kabra, M.; Gupta, N. Bi-allelic loss-of-function novel variants in LTBP3-related skeletal dysplasia: Report of first patient from India. Am. J. Med. Genet. 2020, 182, 1944–1946. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortüm, F.; Caputo, V.; Bauer, C.K.; Stella, L.; Ciolfi, A.; Alawi, M.; Bocchinfuso, G.; Flex, E.; Paolacci, S.; Dentici, M.L.; et al. Mutations in KCNH1 and ATP6V1B2 cause Zimmermann-Laband syndrome. Nat. Genet. 2015, 47, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Flex, E.; Niceta, M.; Cecchetti, S.; Thiffault, I.; Au, M.; Capuano, A.; Piermarini, E.; Ivanova, A.; Francis, J.; Chillemi, G.; et al. Biallelic Mutations in TBCD, Encoding the Tubulin Folding Cofactor D, Perturb Microtubule Dynamics and Cause Early-Onset Encephalopathy. Am. J. Hum. Genet. 2016, 99, 962–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flex, E.; Martinelli, S.; Van Dijck, A.; Ciolfi, A.; Au, M.G.; Capuano, A.; Piermarini, E.; Ivanova, A.A.; Francis, J.W.; Chillemi, G.; et al. Aberrant Function of the C-Terminal Tail of HIST1H1E Accelerates Cellular Senescence and Causes P remature Aging. Am. J. Hum. Genet. 2019, 105, 493–508. [Google Scholar] [CrossRef] [Green Version]
- Bauer, C.K.; Calligari, P.; Radio, F.C.; Caputo, V.; Dentici, M.L.; Falah, N.; High, F.; Pantaleoni, F.; Barresi, S.; Ciolfi, A.; et al. Mutations in KCNK4 that Affect Gating Cause a Recognizable Neurodevelopmental Syndrome. Am. J. Hum. Genet. 2018, 103, 621–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wangle, L.; Coon, M.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP v2.0: A database of human non-synonymous SNVs and their functional predictions and annotations. Hum. Mutat. 2013, 34, E2393–E2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagadeesh, K.; Wenger, A.; Berger, M.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants with uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef]
- Li, Q.; Wang, K. InterVar: Clinical interpretation of genetic variants by ACMG-AMP 2015 guideline. Am. J. Hum. Genet. 2017, 100, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Dendunnen, J.T.; Antonarakis, S.E. Mutation nomenclature extensions and suggestions to describe complex mutations: A discussion. Hum. Mutat. 2000, 15, 7–12. [Google Scholar] [CrossRef]
- Kanzaki, T.; Olofsson, A.; Moren, A.; Wernstedt, C.; Hellman, U.; Miyazono, K.; Claesson-Welsh, L.; Heldin, C.H. TGF-β 1 binding protein: A component of the large latent complex of TGF-β 1 with multiple repeat sequences. Cell 1990, 61, 1051–1061. [Google Scholar] [CrossRef]
- Yin, W.; Smiley, E.; Germiller, J.; Mecham, R.P.; Florer, J.B.; Wenstrup, R.J.; Bonadio, J. Isolation of a novel latent transforming growth factor-β binding protein gene (LTBP-3). J. Biol. Chem. 1995, 270, 10147–10160. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Dabovic, B.; Annes, J.P.; Rifkin, D.B. Latent TGF-β binding protein-3 (LTBP-3) requires binding to TGF-β for secretion. FEBS Lett. 2002, 517, 277–280. [Google Scholar] [CrossRef] [Green Version]
- Penttinen, C.; Saharinen, J.; Weikkolainen, K.; Hyytiainen, M.; Keski-Oja, J. Secretion of human latent TGF-β-binding protein-3 (LTBP-3) is dependent on co-expression of TGF-β. J. Cell Sci. 2002, 115, 3457–3468. [Google Scholar] [CrossRef]
- Dabovic, B.; Chen, Y.; Colarossi, C.; Zambuto, L.; Obata, H.; Rifkin, D.B. Bone defects in latent TGF-β binding protein (Ltbp)-3 null mice; a role for Ltbp in TGF-β presentation. J. Endocrinol. 2002, 175, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Dabovic, B.; Levasseur, R.; Zambuto, L.; Chen, Y.; Karsenty, G.; Rifkin, D.B. Osteopetrosis-like phenotype in latent TGF-β binding protein 3 deficient mice. Bone 2005, 37, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K.; Olofsson, A.; Colosetti, P.; Heldin, C.H. A role of the latent TGF-β 1-binding protein in the assembly and secretion of TGF-β 1. EMBO J. 1991, 10, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K.; Thyberg, J.; Heldin, C.H. Retention of the transforming growth factor-β 1 precursor in the Golgi complex in a latent endoglycosidase H-sensitive form. J. Biol. Chem. 1992, 267, 5668–5675. [Google Scholar] [CrossRef]
- Taipale, J.; Miyazono, K.; Heldin, C.H.; Keski-Oja, J. Latent transforming growth factor-β 1 associates to fibroblast extracellular matrix via latent TGF-β binding protein. J. Cell Biol. 1994, 124, 171–181. [Google Scholar] [CrossRef]
- Robertson, I.B.; Horiguchi, M.; Zilberberg, L.; Dabovic, B.; Hadjiolova, K.; Rifkin, D.B. Latent TGF-β-binding proteins. Matrix Biol. 2015, 47, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Finocchiaro, L.M.; Arzt, E.S.; Fernández-Castelo, S.; Criscuolo, M.; Finkielman, S.; Nahmod, V.E. Serotonin and melatonin synthesis in peripheral blood mononuclear cells: Stimulation by interferon-γ as part of an immunomodulatory pathway. J. Interferon Res. 1988, 8, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.C.; Bagade, S.; McQueen, M.B.; Ioannidis, J.P.A.; Kavvoura, F.K.; Khoury, M.J.; Tanzi, R.E.; Bertram, L. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: The SzGene database. Nat. Genet. 2008, 40, 827–834. [Google Scholar] [CrossRef]
- Li, D.; He, L. Further clarification of the contribution of the tryptophan hydroxylase (TPH) gene to suicidal behavior using systematic allelic and genotypic meta-analyses. Hum. Genet 2006, 119, 233–240. [Google Scholar] [CrossRef]
- Al-Ani, A.H.; Antoun, J.S.; Thomson, W.M.; Topless, R.; Merriman, T.R.; Farella, M. Common variants of EDA are associated with non-syndromic hypodontia. Orthod. Craniofac. Res. 2021, 24, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Galluccio, G.; Castellano, M.; La Monaca, C. Genetic basis of non-syndromic anomalies of human tooth number. Arch. Oral Biol. 2012, 57, 918–930. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Location | Gene | Transcript | Variation | Protein Level | Note | |
|---|---|---|---|---|---|---|
| Homozygous | chr11:65308427 | LTBP3 | NM_001130144.3 | c.2894-2A>G | p.Ala965Glyfs*38 | Latent transforming growth factor (TGF) beta binding protein |
| chr11:18044351 | TPH1 | NM_004179.3 | c.1154A>T | p. Lys385Met | Tryptophan hydroxylase 1 | |
| Hemizygous | chrX:69255334 | EDA | NM_001399.5 | c.1051G>A | p.Val351Ile | Ectodysplasin A |
| Noor et al., (2009) | Huckert et al., (2015) | Dugan et al., (2015) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| General Information | Age (y/m) | 30y a | 14y | 13y b | 13y b | 11y | 16y c,9y c,12y c | 18y d | 15y d | |
| Gender (M/F) | M | F | F | M | M | F,F,M | F | F | ||
| Number of affected individuals studied in the family | 4 | 2 | 2 | 1 | 3 | 2 | ||||
| Ethnic | Punjabi | Turkey | Caucasian French | Brazil | Pakistan | Emirati | ||||
| Parental conseguinity | Yes | Yes | No | Yes | Yes | No | ||||
| Clinical Features | Growth | Normal birth length | NA | NA | NA | NA | NA | NA | − | + |
| Short stature | + | + | + | + | + | + | + | + | ||
| Short neck | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Short trunk | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Eyes | Corneal Opacities | NA | NA | NA | NA | NA | NA | NA | NA | |
| Myopia | NA | + | − | − | NA | NA | NA | NA | ||
| Hyperopia | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Teeth | Retarded teeth eruption | NA | NA | NA | NA | + | NA | + | + | |
| Amelogenesis imperfecta | + | + | + | + | + | + | NA | NA | ||
| Oligodotia | + | + | NA | NA | − | + | + | + | ||
| Skeletal | Pectus carinatum | NA | NA | NA | + | NA | NA | NA | NA | |
| Osteopenia | − | + | NA | NA | NA | + | NA | NA | ||
| Platyspondyly | − | + | + | NA | + | + | NA | NA | ||
| Short pedicles | NA | NA | NA | NA | NA | NA | + | + | ||
| Vertebral borders rounded anteriorly and posteriorly | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Irregular end plates | + | + | ||||||||
| Narrow intervertebral spaces | NA | NA | NA | NA | NA | NA | + | + | ||
| Scoliosis | + | + | NA | NA | + | + | + | + | ||
| Kyphosis | NA | NA | NA | NA | NA | NA | + | + | ||
| Gibbus | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Short femoral neck | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Irregular femoral metaphyses | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Short iliac bones | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Irregular epiphyses | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Short and bowed lower limbs | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Enlarged knee joints | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Metaphyseal changes of knees and hips | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Osteoarthropathy, precocious | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Slightly short long bones | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Skin, Hands & Hair | Brachydactyly | − | − | + | + | NA | NA | ± | ± | |
| Arachnodactyly | NA | NA | NA | NA | NA | NA | + | − | ||
| Acne | NA | NA | NA | NA | NA | NA | − | − | ||
| Acanthosis nigricans | NA | NA | NA | NA | NA | NA | + | − | ||
| Hirsutism | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Neurologic | Normal intelligence | + | NA | NA | NA | NA | NA | + | − | |
| Spinal cord compression | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Falx cerebri, precocious calcification of | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Cardiac | Thoracic aortic aneurysms/dissections | NA | NA | NA | NA | NA | NA | NA | NA | |
| Mild mitral valve prolapse | NA | NA | NA | NA | NA | NA | + | + | ||
| Cardiomyopathy | NA | − | − | − | NA | NA | + | + | ||
| Molecular Finding | Inheritance patterns | AR | AR | AR* | AR* | AR | AR | AR | AR | |
| Gene | LTBP3 | LTBP3 | LTBP3 | LTBP3 | LTBP3 | LTBP3 | LTBP3 | LTBP3 | ||
| Exon | 16 | 14 | 2/8 | 2/8 | 15 | 17 | 13 | 13 | ||
| DNA | c.2322C>G | c.2071_2084delTAC CGG CTC AAA GC | c.421C>T; c.1531+1G>T | c.421C>T; c.1531+1G>T | c.2216_2217delG | c.2356_2357delG | c.1858_1859delG | c.1858_1859delG | ||
| Protein | p.Tyr744* | p.Tyr691Leufs*95 | p.Gln141*; p.(?) | p.Gln141*; p.(?) | p.Gly739Alafs*7 | p.Val786Trpfs*82 | p.Cys620Trpfs*171 | p.Cys620Trpfs*171 | ||
| Guo et al., (2018) | Intarak et al., (2019) | Kaur et al., (2020) | Present Study | |||||||
| General Information | Age (y/m) | 54y e | 55y e | 59y e | 44y f | 58y f | 24y g | 7y | 14y | |
| Gender (M/F) | M | F | F | M | F | M | F | M | ||
| Number of affected individuals studied in the family | 3 | 2 | 1 | 1 | 1 | |||||
| Ethnic | American | American | Thai | India | Perù | |||||
| Parental conseguinity | No | Yes | Yes | No | Yes | |||||
| Clinical Features | Growth | Normal birth length | NA | NA | NA | NA | NA | NA | + | + |
| Short stature | + | + | + | + | + | + | + | + | ||
| Short neck | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Short trunk | NA | NA | NA | NA | NA | NA | + | + | ||
| Eyes | Corneal Opacities | NA | NA | NA | NA | NA | NA | NA | − | |
| Myopia | NA | NA | NA | NA | NA | − | NA | − | ||
| Hyperopia | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Teeth | Retarded teeth eruption | NA | NA | NA | NA | NA | − | + | NA | |
| Amelogenesis imperfecta | + | + | + | + | + | + | + | + | ||
| Oligodotia | NA | NA | NA | NA | NA | − | + | + | ||
| Skeletal | Pectus carinatum | NA | NA | NA | NA | NA | − | NA | − | |
| Osteopenia | − | − | + | − | + | NA | − | − | ||
| Platyspondyly | NA | NA | NA | NA | NA | NA | + | + | ||
| Short pedicles | NA | NA | NA | NA | NA | NA | NA | |||
| Vertebral borders rounded anteriorly and posteriorly | NA | NA | NA | NA | NA | NA | NA | − | ||
| Irregular end plates | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Narrow intervertebral spaces | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Scoliosis | − | − | + | − | + | + | − | + | ||
| Kyphosis | NA | NA | NA | NA | NA | NA | NA | − | ||
| Gibbus | NA | NA | NA | NA | NA | NA | NA | − | ||
| Short femoral neck | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Irregular femoral metaphyses | NA | NA | NA | NA | NA | NA | NA | + | ||
| Short iliac bones | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Irregular epiphyses | NA | NA | NA | NA | NA | NA | + | + | ||
| Short and bowed lower limbs | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Enlarged knee joints | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Metaphyseal changes of knees and hips | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Osteoarthropathy, precocious | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Slightly short long bones | NA | NA | NA | NA | NA | NA | + | NA | ||
| Skin, Hands & hair | Brachydactyly | NA | NA | NA | NA | NA | NA | + | + | |
| Arachnodactyly | NA | NA | NA | NA | NA | NA | − | − | ||
| Acne | NA | NA | NA | NA | NA | NA | NA | − | ||
| Acanthosis nigricans | NA | NA | NA | NA | NA | NA | NA | − | ||
| Hirsutism | NA | NA | NA | NA | NA | NA | NA | − | ||
| Neurologic | Normal intelligence | NA | NA | NA | NA | NA | NA | NA | + | |
| Spinal cord compression | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Falx cerebri, precocious calcification of | NA | NA | NA | NA | NA | NA | NA | NA | ||
| Cardiac | Thoracic aortic aneurysms/dissections | + | + | − | + | + | − | − | NA | |
| Mild mitral valve prolapse | − | + | + | − | + | ? | − | NA | ||
| Cardiomyopathy | NA | NA | NA | NA | NA | NA | − | + | ||
| Molecular Finding | Inheritance patterns | AR* | AR* | AR* | AR | AR | AR | AR* | AR | |
| Gene | LTBP3 | LTBP3 | LTBP3 | LTBP3 | LTBP3 | LTBP3 | LTBP3 | LTBP3 | ||
| Exon | 1/16 | 1/16 | 1/16 | 14 | 14 | splice site acceptor before exon 12 | splice site acceptor before exon 21 | |||
| DNA | c.132delG; c.2248G>T | c.132delG; c.2248G>T | c.132delG; c.2248G>T | c.2033_2041delinsCTT | c.2033_2041delinsCTT | c.1721-2A>G | c.3153_3154del; c.689_690del | c.2894-2A>G | ||
| Protein | p.Pro45Argfs*25; p.Glu750* | p.Pro45Argfs*25; p.Glu750* | p.Pro45Argfs*25; p.Glu750* | p.Asn678_Gly681delinsThrCys | p.Asn678_Gly681delinsThrCys | p.? | p.Cys1051*; p.Val230Alafs*16 | p.(Ala965Glyfs*38) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flex, E.; Imperatore, V.; Carpentieri, G.; Bruselles, A.; Ciolfi, A.; Pizzi, S.; Tedesco, M.G.; Rogaia, D.; Mencarelli, A.; Di Cara, G.; et al. A Rare Case of Brachyolmia with Amelogenesis Imperfecta Caused by a New Pathogenic Splicing Variant in LTBP3. Genes 2021, 12, 1406. https://doi.org/10.3390/genes12091406
Flex E, Imperatore V, Carpentieri G, Bruselles A, Ciolfi A, Pizzi S, Tedesco MG, Rogaia D, Mencarelli A, Di Cara G, et al. A Rare Case of Brachyolmia with Amelogenesis Imperfecta Caused by a New Pathogenic Splicing Variant in LTBP3. Genes. 2021; 12(9):1406. https://doi.org/10.3390/genes12091406
Chicago/Turabian StyleFlex, Elisabetta, Valentina Imperatore, Giovanna Carpentieri, Alessandro Bruselles, Andrea Ciolfi, Simone Pizzi, Maria Giovanna Tedesco, Daniela Rogaia, Amedea Mencarelli, Giuseppe Di Cara, and et al. 2021. "A Rare Case of Brachyolmia with Amelogenesis Imperfecta Caused by a New Pathogenic Splicing Variant in LTBP3" Genes 12, no. 9: 1406. https://doi.org/10.3390/genes12091406
APA StyleFlex, E., Imperatore, V., Carpentieri, G., Bruselles, A., Ciolfi, A., Pizzi, S., Tedesco, M. G., Rogaia, D., Mencarelli, A., Di Cara, G., Verrotti, A., Troiani, S., Merla, G., Tartaglia, M., & Prontera, P. (2021). A Rare Case of Brachyolmia with Amelogenesis Imperfecta Caused by a New Pathogenic Splicing Variant in LTBP3. Genes, 12(9), 1406. https://doi.org/10.3390/genes12091406