
Candidate Genes and Pathways Associated with Gilles de la Tourette Syndrome—Where Are We?



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Candidate Gene and Pathway Studies
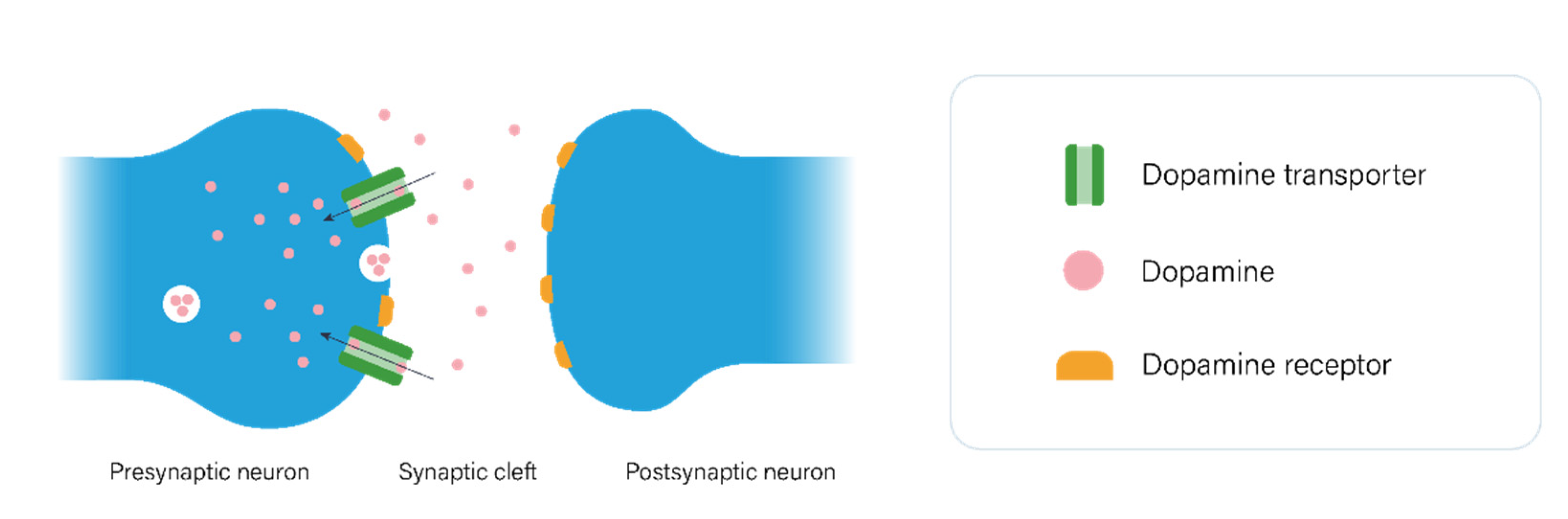
2.1. The Dopaminergic Pathway
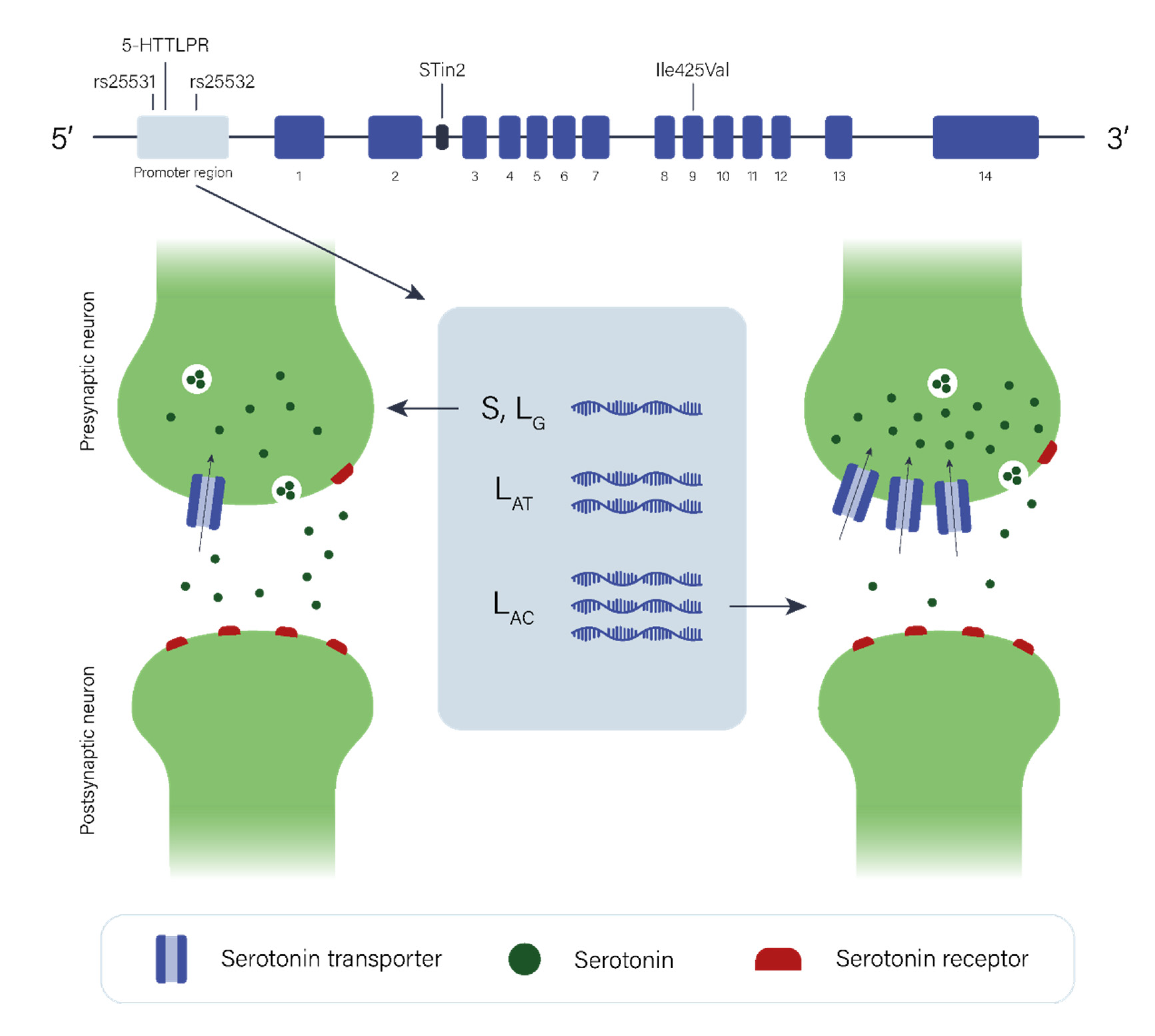
2.2. The Serotonergic Pathway
3. Linkage Analyses, GWAS & Other Studies
3.1. SLITRK1
3.2. IMMPL2
3.3. HDC
3.4. CELSR3, WWC1, FN1, and NIPBL
3.5. ASH1L
3.6. FLT3
3.7. NRXN1 and CNTN6
4. Discussion—Future Opportunities and Challenges
5. Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- Knight, T.; Steeves, T.; Day, L.; Lowerison, M.; Jette, N.; Pringsheim, T. Prevalence of Tic Disorders: A Systematic Review and Meta-Analysis. Pediatr. Neurol. 2012, 47, 77–90. [Google Scholar] [CrossRef]
- Scharf, J.M.; Miller, L.L.; Bs, C.A.G.; Ma, J.A.; Mathews, C.A.; Ben-Shlomo, Y. Population prevalence of Tourette syndrome: A systematic review and meta-analysis. Mov. Disord. 2014, 30, 221–228. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- Müller-Vahl, K.R.; Sambrani, T.; Jakubovski, E. Tic disorders revisited: Introduction of the term “tic spectrum disorders”. Eur. Child Adolesc. Psychiatry 2019, 28, 1129–1135. [Google Scholar] [CrossRef]
- Conte, G.; Valente, F.; Fioriello, F.; Cardona, F. Rage attacks in Tourette Syndrome and Chronic Tic Disorder: A systematic review. Neurosci. Biobehav. Rev. 2020, 119, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Darrow, S.M.; Grados, M.; Sandor, P.; Hirschtritt, M.E.; Illmann, C.; Osiecki, L.; Dion, Y.; King, R.; Pauls, D.; Budman, C.L.; et al. Autism Spectrum Symptoms in a Tourette’s Disorder Sample. J. Am. Acad. Child Adolesc. Psychiatry 2017, 56, 610–617.e1. [Google Scholar] [CrossRef] [PubMed]
- Huisman-van Dijk, H.M.; van de Schoot, R.; Rijkeboer, M.M.; Mathews, C.A.; Cath, D.C. The relationship between tics, OC, ADHD and autism symptoms: A cross- disorder symptom analysis in Gilles de la Tourette syndrome patients and family-members. Psychiatry Res. 2016, 237, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; García-Martín, E.; Agundez, J. Sleep disorders in tourette syndrome. Sleep Med. Rev. 2020, 53, 101335. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.M.; Eapen, V.; Singer, H.S.; Martino, D.; Scharf, J.M.; Paschou, P.; Roessner, V.; Woods, D.W.; Hariz, M.; Mathews, C.A.; et al. Gilles de la Tourette syndrome. Nat. Rev. Dis. Prim. 2017, 3, 16097. [Google Scholar] [CrossRef]
- Vermilion, J.; Pedraza, C.; Augustine, E.F.; Adams, H.R.; Vierhile, A.; Lewin, A.B.; Collins, A.T.; McDermott, M.P.; O’Connor, T.; Kurlan, R.; et al. Anxiety Symptoms Differ in Youth With and Without Tic Disorders. Child Psychiatry Hum. Dev. 2021, 52, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, P.J.; Dietrich, A.; Edwards, M.J.; Elamin, I.; Martino, D. Environmental factors in Tourette syndrome. Neurosci. Biobehav. Rev. 2013, 37, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Frick, L.; Pittenger, C. Microglial Dysregulation in OCD, Tourette Syndrome, and PANDAS. J. Immunol. Res. 2016, 2016, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Paschou, P. The genetic basis of Gilles de la Tourette Syndrome. Neurosci. Biobehav. Rev. 2013, 37, 1026–1039. [Google Scholar] [CrossRef]
- Draganski, B.; Martino, D.; Cavanna, A.E.; Hutton, C.; Orth, M.; Robertson, M.M.; Critchley, H.D.; Frackowiak, R.S. Multispectral brain morphometry in Tourette syndrome persisting into adulthood. Brain 2010, 133, 3661–3675. [Google Scholar] [CrossRef]
- Peterson, B.S.; Thomas, P.; Kane, M.J.; Scahill, L.; Zhang, H.; Bronen, R.; King, R.A.; Leckman, J.F.; Staib, L. Basal Ganglia Volumes in Patients With Gilles de la Tourette Syndrome. Arch. Gen. Psychiatry 2003, 60, 415–424. [Google Scholar] [CrossRef]
- Wang, Z.; Maia, T.; Marsh, R.; Colibazzi, T.; Gerber, A.; Peterson, B.S. The Neural Circuits That Generate Tics in Tourette’s Syndrome. Am. J. Psychiatry 2011, 168, 1326–1337. [Google Scholar] [CrossRef] [PubMed]
- Neuner, I.; Schneider, F.; Shah, N.J. Functional Neuroanatomy of Tics. In International Review of Neurobiology; Harris, R.A., Jenner, P., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; pp. 35–71. [Google Scholar] [CrossRef]
- Cumming, P. The life history of dopamine. In Imaging Dopamine; Cambridge University Press: Cambridge, UK, 2009; pp. 5–18. [Google Scholar]
- Beaulieu, J.-M.; Gainetdinov, R. The Physiology, Signaling, and Pharmacology of Dopamine Receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.; Zametkin, A.J.; Jons, P.H.; Matochik, J.A.; Pascualvaca, D.; Cohen, R.M. High Presynaptic Dopaminergic Activity in Children With Tourette’s Disorder. J. Am. Acad. Child Adolesc. Psychiatry 1999, 38, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Lavenstein, B.L. Treatment approaches for children with Tourette’s syndrome. Curr. Neurol. Neurosci. Rep. 2003, 3, 143–148. [Google Scholar] [CrossRef]
- Swain, J.; Scahill, L.; Lombroso, P.J.; King, R.A.; Leckman, J.F. Tourette Syndrome and Tic Disorders: A Decade of Progress. J. Am. Acad. Child Adolesc. Psychiatry 2007, 46, 947–968. [Google Scholar] [CrossRef]
- Comings, D.E.; Comings, B.G.; Muhleman, D.; Dietz, G.; Shahbahrami, B.; Tast, D.; Knell, E.; Kocsis, P.; Baumgarten, R.; Kovacs, B.W.; et al. The Dopamine D2 Receptor Locus as a Modifying Gene in Neuropsychiatric Disorders. JAMA 1991, 266, 1793–1800. [Google Scholar] [CrossRef]
- Comings, D.; Muhleman, D.; Dietz, G.; Dino, M.; Legro, R.; Gade, R. Association between Tourette’s syndrome and homozygosity at the dopamine D3 receptor gene. Lancet 1993, 341, 906. [Google Scholar] [CrossRef]
- Comings, D.E.; Wu, S.; Chiu, C.; Ring, R.; Gade, R.; Ahn, C.; MacMurray, J.P.; Dietz, G.; Muhleman, D. Polygenic inheritance of Tourette syndrome, stuttering, attention deficit hyperactivity, conduct, and oppositional defiant disorder: The additive and subtractive effect of the three dopaminergic genes—DRD2, DβH, and DAT1. Am. J. Med. Genet. 1996, 67, 264–288. [Google Scholar] [CrossRef]
- Cruz, C.; Camarena, B.; King, N.; Páez, F.; Sidenberg, D.; De La Fuente, J.R.; Nicolini, H. Increased prevalence of the seven-repeat variant of the dopamine D4 receptor gene in patients with obsessive-compulsive disorder with tics. Neurosci. Lett. 1997, 231, 1–4. [Google Scholar] [CrossRef]
- Gade, R.; Muhleman, D.; Blake, H.; MacMurray, J.; Johnson, P.; Verde, R.; Saucier, G.; Comings, D.E. Correlation of length of VNTR alleles at the X-linked MAOA gene and phenotypic effect in Tourette syndrome and drug abuse. Mol. Psychiatry 1998, 3, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Grice, D.E.; Leckman, J.F.; Pauls, D.L.; Kurlan, R.; Kidd, K.K.; Pakstis, A.J.; Chang, F.M.; Buxbaum, J.; Cohen, D.J.; Gelernter, J. Linkage disequilibrium between an allele at the dopamine D4 receptor locus and Tourette syndrome, by the transmission-disequilibrium test. Am. J. Hum. Genet. 1996, 59, 644–652. [Google Scholar] [PubMed]
- Rowe, D.C.; Stever, C.; Gard, J.M.C.; Cleveland, H.H.; Sanders, M.L.; Abramowitz, A.; Kozol, S.T.; Mohr, J.H.; Sherman, S.L.; Waldman, I.D. The relation of the dopamine transporter gene (DAT1) to symptoms of internalizing disorders in children. Behav. Genet. 1998, 28, 215–225. [Google Scholar] [CrossRef]
- Díaz-Anzaldúa, A.; Joober, R.; Rivière, J.B.; Dion, Y.; Lespérance, P.; Richer, F.; Chouinard, S.; Rouleau, G.A. Tourette syndrome and dopaminergic genes: A family-based association study in the French Canadian founder population. Mol. Psychiatry 2004, 9, 272–277. [Google Scholar] [CrossRef]
- Huertas-Fernández, I.; Gómez-Garre, P.; Madruga-Garrido, M.; Bernal, I.; Bonilla-Toribio, M.; Martín-Rodríguez, J.F.; Cáceres-Redondo, M.T.; Vargas-González, L.; Carrillo, F.; Pascual, A.; et al. GDNF gene is associated with tourette syndrome in a family study. Mov. Disord. 2015, 30, 1115–1120. [Google Scholar] [CrossRef]
- Tárnok, Z.; Rónai, Z.; Gervai, J.; Kereszturi, E.; Gadoros, J.; Sasvari-Szekely, M.; Nemoda, Z. Dopaminergic candidate genes in Tourette syndrome: Association between tic severity and 3′ UTR polymorphism of the dopamine transporter gene. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2007, 144B, 900–905. [Google Scholar] [CrossRef]
- Yoon, D.Y.; Rippel, C.A.; Kobets, A.J.; Morris, C.M.; Lee, J.E.; Williams, P.N.; Bridges, D.D.; Vandenbergh, D.; Shugart, Y.Y.; Singer, H.S. Dopaminergic polymorphisms in Tourette syndrome: Association with the DAT gene (SLC6A3). Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2007, 144B, 605–610. [Google Scholar] [CrossRef]
- Liu, S.; Cui, J.; Zhang, X.; Wu, W.; Niu, H.; Ma, X.; Xu, H.; Yi, M. Variable number tandem repeats in dopamine receptor D4 in Tourette’s syndrome. Mov. Disord. 2014, 29, 1687–1691. [Google Scholar] [CrossRef] [PubMed]
- Yuan, A.; Su, L.; Yu, S.; Li, C.; Yu, T.; Sun, J. Association between DRD2/ANKK1 TaqIA Polymorphism and Susceptibility with Tourette Syndrome: A Meta-Analysis. PLoS ONE 2015, 10, e0131060. [Google Scholar] [CrossRef] [PubMed]
- Ponce, G.; Lombraña, A.Q.; Martín-Palanco, N.G.; Rubio-Solsona, E.; Jimenez-Arriero, M.A.; Palomo, T.; Hoenicka, J. The Addiction-Related Gene Ankk1 is Oppositely Regulated by D1R- and D2R-Like Dopamine Receptors. Neurotox. Res. 2015, 29, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Ponce, G.; Pérez-González, R.; Aragüés, M.; Palomo, T.; Rodríguez-Jiménez, R.; Jiménez-Arriero, M.A.; Hoenicka, J. The ANKK1 Kinase Gene and Psychiatric Disorders. Neurotox. Res. 2009, 16, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Müller-Vahl, K.R.; Loeber, G.; Kotsiari, A.; Müller-Engling, L.; Frieling, H. Gilles de la Tourette syndrome is associated with hypermethylation of the dopamine D2 receptor gene. J. Psychiatr. Res. 2017, 86, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sinopoli, V.M.; Burton, C.; Kronenberg, S.; Arnold, P.D. A review of the role of serotonin system genes in obsessive-compulsive disorder. Neurosci. Biobehav. Rev. 2017, 80, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Olivier, B. Serotonin: A never-ending story. Eur. J. Pharmacol. 2015, 753, 2–18. [Google Scholar] [CrossRef]
- Anderson, G.M.; Pollak, E.S.; Chatterjee, D.; Leckman, J.F.; Riddle, M.A.; Cohen, D.J. Brain Monoamines and Amino Acids in Gilles de la Tourette’s Syndrome: A Preliminary Study of Subcortical Regions. Arch. Gen. Psychiatry 1992, 49, 584–586. [Google Scholar] [CrossRef]
- Butler, I.J.; Koslow, S.H.; Seifert, W.E.; Caprioli, R.M.; Singer, H.S. Biogenic amine metabolism in tourette syndrome. Ann. Neurol. 1979, 6, 37–39. [Google Scholar] [CrossRef]
- Leckman, J.F.; Anderson, G.M.; Cohen, D.J.; Ort, S.; Harcherik, D.F.; Hoder, E.; Shaywitz, B.A. Whole blood serotonin and tryptophan levels in Tourette’s disorder: Effects of acute and chronic clonidine treatment. Life Sci. 1984, 35, 2497–2503. [Google Scholar] [CrossRef]
- Leckman, J.F.; Goodman, W.K.; Anderson, G.M.; Riddle, M.A.; Chappell, P.B.; McSwiggan-Hardin, M.T.; McDougle, C.J.; Scahill, L.D.; Ort, S.I.; Pauls, D.L.; et al. Cerebrospinal Fluid Biogenic Amines in Obsessive Compulsive Disorder, Tourette’s Syndrome, and Healthy Controls. Neuropsychopharmacology 1995, 12, 73–86. [Google Scholar] [CrossRef][Green Version]
- Müller-Vahl, K.R.; Meyer, G.J.; Knapp, W.H.; Emrich, H.M.; Gielow, P.; Brücke, T.; Berding, G. Serotonin transporter binding in Tourette Syndrome. Neurosci. Lett. 2005, 385, 120–125. [Google Scholar] [CrossRef]
- Steeves, T.D.; Fox, S.H. Neurobiological Basis of Serotonin-Dopamine Antagonists in the Treatment of Gilles de La Tourette Syndrome. Prog. Brain Res. 2008, 172, 495–513. [Google Scholar] [PubMed]
- Bortolozzi, A.; Diaz-Mataix, L.; Scorza, M.C.; Celada, P.; Artigas, F. The activation of 5-HT2A receptors in prefrontal cortex enhances dopaminergic activity. J. Neurochem. 2005, 95, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- De Deurwaerdère, P.; Navailles, S.; Berg, K.A.; Clarke, W.P.; Spampinato, U. Constitutive Activity of the Serotonin2C Receptor Inhibits In Vivo Dopamine Release in the Rat Striatum and Nucleus Accumbens. J. Neurosci. 2004, 24, 3235–3241. [Google Scholar] [CrossRef] [PubMed]
- Esposito, E.; Di Matteo, V.; Di Giovanni, G. Serotonin–dopamine interaction: An overview. Prog. Brain Res. 2008, 172, 3–6. [Google Scholar] [CrossRef]
- Larsen, M.B.; Sonders, M.S.; Mortensen, O.V.; Larson, G.A.; Zahniser, N.R.; Amara, S.G. Dopamine Transport by the Serotonin Transporter: A Mechanistically Distinct Mode of Substrate Translocation. J. Neurosci. 2011, 31, 6605–6615. [Google Scholar] [CrossRef] [PubMed]
- Dehning, S.; Müller, N.; Matz, J.; Bender, A.; Kerle, I.; Benninghoff, J.; Musil, R.; Spellmann, I.; Bondy, B.; Möller, H.-J.; et al. A genetic variant of HTR2C may play a role in the manifestation of Tourette syndrome. Psychiatr. Genet. 2010, 20, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Su, L.; Zhang, J.; Lei, J.; Deng, X.; Xu, H.; Yang, Z.; Kuang, S.; Tang, J.; Luo, Z.; et al. Analysis of the BTBD9 and HTR2C variants in Chinese Han patients with Tourette syndrome. Psychiatr. Genet. 2012, 22, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Niesler, B.; Frank, B.; Hebebrand, J.; Rappold, G. Serotonin receptor genes HTR3A and HTR3B are not involved in Gilles de la Tourette syndrome. Psychiatr. Genet. 2005, 15, 303–304. [Google Scholar] [CrossRef]
- Moya, P.; Wendland, J.R.; Rubenstein, L.M.; Timpano, K.; Heiman, G.; Tischfield, J.; King, R.A.; Andrews, A.; Ramamoorthy, S.; McMahon, F.; et al. Common and rare alleles of the serotonin transporter gene, SLC6A4, associated with Tourette’s disorder. Mov. Disord. 2013, 28, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Hildonen, M.; Levy, A.M.; Dahl, C.; Bjerregaard, V.A.; Møller, L.B.; Guldberg, P.; Debes, N.M.; Tümer, Z. Elevated Expression of SLC6A4 Encoding the Serotonin Transporter (SERT) in Gilles de la Tourette Syndrome. Genes 2021, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Cavallini, M.C.; Di Bella, D.; Catalano, M.; Bellodi, L. An association study between 5-HTTLPR polymorphism, COMT polymorphism, and Tourette’s syndrome. Psychiatry Res. 2000, 97, 93–100. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, X.; Yin, Y.; Wang, M.; Che, F.; Ma, X. An Association Analysis between 5-HTTLPR Polymorphism and Obsessive-Compulsive Disorder, Tourette Syndrome in a Chinese Han Population. CNS Neurosci. Ther. 2011, 17, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.-Z.; Lipsky, R.; Zhu, G.; Akhtar, L.A.; Taubman, J.; Greenberg, B.D.; Xu, K.; Arnold, P.D.; Richter, M.A.; Kennedy, J.L.; et al. Serotonin Transporter Promoter Gain-of-Function Genotypes Are Linked to Obsessive-Compulsive Disorder. Am. J. Hum. Genet. 2006, 78, 815–826. [Google Scholar] [CrossRef]
- Cao, X.; Zhang, Y.; Abdulkadir, M.; Deng, L.; Fernandez, T.V.; Julie, B.G.; Pieter, H.; Robert, J.H.; Justin, A.K.; Kuperman, S.; et al. Whole-exome sequencing identifies genes associated with Tourette’s disorder in multiplex families. Mol. Psychiatry 2021, 1–15. [Google Scholar] [CrossRef]
- Mössner, R.; Müller-Vahl, K.; Döring, N.; Stuhrmann, M. Role of the novel tryptophan hydroxylase-2 gene in Tourette syndrome. Mol. Psychiatry 2007, 12, 617–619. [Google Scholar] [CrossRef]
- Zheng, P.; Li, E.; Wang, J.; Cui, X.; Wang, L. Involvement of tryptophan hydroxylase 2 gene polymorphisms in susceptibility to tic disorder in Chinese Han population. Behav. Brain Funct. 2013, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Abdulkadir, M.; Londono, D.; Gordon, D.; Fernandez, T.V.; Brown, L.W.; Cheon, K.-A.; Coffey, B.J.; Elzerman, L.; Fremer, C.; Fründt, O.; et al. Investigation of previously implicated genetic variants in chronic tic disorders: A transmission disequilibrium test approach. Eur. Arch. Psychiatry Clin. Neurosci. 2017, 268, 301–316. [Google Scholar] [CrossRef]
- Abelson, J.F.; Kwan, K.Y.; O’Roak, B.J.; Baek, D.Y.; Stillman, A.A.; Morgan, T.M.; Mathews, C.A.; Pauls, D.L.; Rašin, M.-R.; Gunel, M.; et al. Sequence Variants in SLITRK1 Are Associated with Tourette’s Syndrome. Science 2005, 310, 317–320. [Google Scholar] [CrossRef]
- Deng, H.; Le, W.D.; Xie, W.J.; Jankovic, J. Examination of the SLITRK1 gene in Caucasian patients with Tourette syndrome. Acta Neurol. Scand. 2006, 114, 400–402. [Google Scholar] [CrossRef]
- Keen-Kim, D.; Mathews, C.A.; Reus, V.I.; Lowe, T.L.; Herrera, L.D.; Budman, C.L.; Gross-Tsur, V.; Pulver, A.E.; Bruun, R.D.; Erenberg, G.; et al. Overrepresentation of rare variants in a specific ethnic group may confuse interpretation of association analyses. Hum. Mol. Genet. 2006, 15, 3324–3328. [Google Scholar] [CrossRef]
- Scharf, J.M.; Moorjani, P.; Fagerness, J.; Platko, J.V.; Illmann, C.; Galloway, B.; Jenike, E.; Stewart, S.E.; Pauls, D.L.; Cath, D.; et al. Lack of Association Between Slitrk1var321 and Tourette Syndrome in a Large Family-Based Sample. Neurology 2008, 70, 1495–1496. [Google Scholar] [CrossRef]
- Yasmeen, S.; Melchior, L.; Bertelsen, B.; Skov, L.; Debes, N.; Tümer, Z. Sequence analysis of SLITRK1 for var321 in Danish patients with Tourette syndrome and review of the literature. Psychiatr. Genet. 2013, 23, 130–133. [Google Scholar] [CrossRef]
- Zimprich, A.; Hatala, K.; Riederer, F.; Stogmann, E.; Aschauer, H.N.; Stamenkovic, M. Sequence analysis of the complete SLITRK1 gene in Austrian patients with Tourette’s disorder. Psychiatr. Genet. 2008, 18, 308–309. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Morgan, T.M.; Fishman, D.O.; Saus, E.; Alonso, P.; Gratacòs, M.; Estivill, X.; Teltsh, O.; Kohn, Y.; Kidd, K.K.; et al. Additional support for the association of SLITRK1 var321 and Tourette syndrome. Mol. Psychiatry 2010, 15, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; Potamianou, H.; Xing, J.; Deng, L.; Karagiannidis, I.; Tsetsos, F.; Drineas, P.; Tarnok, Z.; Rizzo, R.; Wolanczyk, T.; et al. Targeted Re-Sequencing Approach of Candidate Genes Implicates Rare Potentially Functional Variants in Tourette Syndrome Etiology. Front. Neurosci. 2016, 10, 428. [Google Scholar] [CrossRef] [PubMed]
- Chou, I.-C.; Wan, L.; Liu, S.-C.; Tsai, C.-H.; Tsai, F.-J. Association of the Slit and Trk-like 1 Gene in Taiwanese Patients With Tourette Syndrome. Pediatr. Neurol. 2007, 37, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Depienne, C.; Ciura, S.; Trouillard, O.; Bouteiller, D.; Leitão, E.; Nava, C.; Keren, B.; Marie, Y.; Guegan, J.; Forlani, S.; et al. Association of Rare Genetic Variants in Opioid Receptors with Tourette Syndrome. Tremor Other Hyperkinet. Mov. (N. Y.) 2019, 9. [Google Scholar] [CrossRef]
- Inai, A.; Tochigi, M.; Kuwabara, H.; Nishimura, F.; Kato, K.; Eriguchi, Y.; Shimada, T.; Furukawa, M.; Kawamura, Y.; Sasaki, T.; et al. Analysis of SLITRK1 in Japanese patients with Tourette syndrome using a next-generation sequencer. Psychiatr. Genet. 2015, 25, 256–258. [Google Scholar] [CrossRef] [PubMed]
- Karagiannidis, I.; Rizzo, R.; Tarnok, Z.; Wolanczyk, T.; Hebebrand, J.; Nöthen, M.M.; Lehmkuhl, G.; Farkas, L.; Nagy, P.; Barta, C.; et al. Replication of association between a SLITRK1 haplotype and Tourette Syndrome in a large sample of families. Mol. Psychiatry 2012, 17, 665–668. [Google Scholar] [CrossRef]
- Miranda, D.M.; Wigg, K.; Kabia, E.M.; Feng, Y.; Sandor, P.; Barr, C.L. Association ofSLITRK1to Gilles de la Tourette Syndrome. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2009, 150B, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Scharf, J.M.; Yu, D.; Mathews, C.A.; Neale, B.M.; Stewart, S.E.; Fagerness, J.A.; Evans, P.; Gamazon, E.; Edlund, C.K.; Service, S.K.; et al. Genome-wide association study of Tourette’s syndrome. Mol. Psychiatry 2013, 18, 721–728. [Google Scholar] [CrossRef]
- Yu, D.; Mathews, C.A.; Scharf, J.M.; Neale, B.M.; Davis, L.K.; Gamazon, E.R.; Derks, E.; Evans, P.; Edlund, C.K.; Crane, J.; et al. Cross-Disorder Genome-Wide Analyses Suggest a Complex Genetic Relationship Between Tourette’s Syndrome and OCD. Am. J. Psychiatry 2015, 172, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Sul, J.H.; Tsetsos, F.; Nawaz, M.S.; Huang, A.Y.; Zelaya, I.; Illmann, C.; Osiecki, L.; Darrow, S.M.; Hirschtritt, M.E.; et al. Interrogating the Genetic Determinants of Tourette’s Syndrome and Other Tic Disorders Through Genome-Wide Association Studies. Am. J. Psychiatry 2019, 176, 217–227. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, X.; Deng, Z.; Li, G.; Zhang, R.; Yang, Z.; Che, F.; Liu, S.; Li, H. The role of SLITRK6 in the pathogenesis of Tourette syndrome: From the conclusion of a family-based study in the Chinese Han population. J. Gene Med. 2020, 22, e3173. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Mathews, C.A.; Stewart, S.E.; Shmelkov, S.V.; Mezey, J.G.; Rodriguez-Flores, J.L.; Rasmussen, S.A.; Britton, J.C.; Oh, Y.-S.; Walkup, J.T.; et al. Rare Synaptogenesis-Impairing Mutations in SLITRK5 Are Associated with Obsessive Compulsive Disorder. PLoS ONE 2017, 12, e0169994. [Google Scholar] [CrossRef]
- Zhang, K.; Feng, Y.; Wigg, K.G.; Sandor, P.; Barr, C. Association study of the SLITRK5 gene and Tourette syndrome. Psychiatr. Genet. 2015, 25, 31–34. [Google Scholar] [CrossRef]
- Carias, K.V.; Wevrick, R. Clinical and genetic analysis of children with a dual diagnosis of Tourette syndrome and autism spectrum disorder. J. Psychiatr. Res. 2019, 111, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Stillman, A.A.; Krsnik, Ž.; Sun, J.; Rašin, M.-R.; State, M.W.; Şestan, N.; Louvi, A. Developmentally regulated and evolutionarily conserved expression of SLITRK1 in brain circuits implicated in Tourette syndrome. J. Comp. Neurol. 2009, 513, 21–37. [Google Scholar] [CrossRef]
- Proenca, C.C.; Gao, K.P.; Shmelkov, S.V.; Rafii, S.; Lee, F.S. Slitrks as emerging candidate genes involved in neuropsychiatric disorders. Trends Neurosci. 2011, 34, 143–153. [Google Scholar] [CrossRef]
- Petek, E.; Windpassinger, C.; Vincent, J.B.; Cheung, J.; Boright, A.P.; Scherer, S.; Kroisel, P.M.; Wagner, K. Disruption of a Novel Gene (IMMP2L) by a Breakpoint in 7q31 Associated with Tourette Syndrome. Am. J. Hum. Genet. 2001, 68, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Boghosian-Sell, L.; Comings, D.E.; Overhauser, J. Tourette syndrome in a pedigree with a 7;18 translocation: Identification of a YAC spanning the translocation breakpoint at 18q22.3. Am. J. Hum. Genet. 1996, 59, 999–1005. [Google Scholar] [PubMed]
- Patel, C.J.; Cooper-Charles, L.; McMullan, D.J.; Walker, J.M.; Davison, V.; Morton, J.E. Translocation breakpoint at 7q31 associated with tics: Further evidence for IMMP2L as a candidate gene for Tourette syndrome. Eur. J. Hum. Genet. 2011, 19, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Anzaldúa, A.; Joober, R.; Rivière, J.-B.; Dion, Y.; Lespérance, P.; Chouinard, S.; Richer, F.; Rouleau, G.A. Association between 7q31 markers and tourette syndrome. Am. J. Med. Genet. Part A 2004, 127A, 17–20. [Google Scholar] [CrossRef]
- Petek, E.; Schwarzbraun, T.; Noor, A.; Patel, M.; Nakabayashi, K.; Choufani, S.; Windpassinger, C.; Stamenkovic, M.; Robertson, M.M.; Aschauer, H.N.; et al. Molecular and genomic studies of IMMP2L and mutation screening in autism and Tourette syndrome. Mol. Genet. Genom. 2006, 277, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Bertelsen, B.; Melchior, L.; Jensen, L.R.; Groth, C.; Glenthøj, B.; Rizzo, R.; Debes, N.M.; Skov, L.; Brøndum-Nielsen, K.; Paschou, P.; et al. Intragenic deletions affecting two alternative transcripts of the IMMP2L gene in patients with Tourette syndrome. Eur. J. Hum. Genet. 2014, 22, 1283–1289. [Google Scholar] [CrossRef] [PubMed]
- Bjerregaard, V.A.; Schönewolf-Greulich, B.; Rasmussen, L.J.; Desler, C.; Tümer, Z. Mitochondrial Function in Gilles de la Tourette Syndrome Patients With and Without Intragenic IMMP2L Deletions. Front. Neurol. 2020, 11, 163. [Google Scholar] [CrossRef]
- Pagliaroli, L.; Vereczkei, A.; Padmanabhuni, S.S.; Tarnok, Z.; Farkas, L.; Nagy, P.; Rizzo, R.; Wolanczyk, T.; Szymanska, U.; Kapisyzi, M.; et al. Association of Genetic Variation in the 3′UTR of LHX6, IMMP2L, and AADAC With Tourette Syndrome. Front. Neurol. 2020, 11, 803. [Google Scholar] [CrossRef]
- Haas, H.L.; Sergeeva, O.A.; Selbach, O. Histamine in the Nervous System. Physiol. Rev. 2008, 88, 1183–1241. [Google Scholar] [CrossRef]
- Ercan-Sencicek, A.G.; Stillman, A.A.; Ghosh, A.K.; Bilguvar, K.; O’Roak, B.J.; Mason, C.E.; Abbott, T.; Gupta, A.; King, R.A.; Pauls, D.L.; et al. L-Histidine Decarboxylase and Tourette’s Syndrome. N. Engl. J. Med. 2010, 362, 1901–1908. [Google Scholar] [CrossRef]
- Karagiannidis, I.; Dehning, S.; Sandor, P.; Tarnok, Z.; Rizzo, R.; Wolanczyk, T.; Madruga-Garrido, M.; Hebebrand, J.; Nöthen, M.; Lehmkuhl, G.; et al. Support of the histaminergic hypothesis in Tourette Syndrome: Association of the histamine decarboxylase gene in a large sample of families. J. Med. Genet. 2013, 50, 760–764. [Google Scholar] [CrossRef]
- Baldan, L.C.; Williams, K.A.; Gallezot, J.-D.; Pogorelov, V.; Rapanelli, M.; Crowley, M.; Anderson, G.M.; Loring, E.; Gorczyca, R.; Billingslea, E.; et al. Histidine Decarboxylase Deficiency Causes Tourette Syndrome: Parallel Findings in Humans and Mice. Neuron 2014, 81, 77–90. [Google Scholar] [CrossRef]
- Fernandez, T.V.; Sanders, S.; Yurkiewicz, I.R.; Ercan-Sencicek, A.G.; Kim, Y.-S.; Fishman, D.O.; Raubeson, M.J.; Song, Y.; Yasuno, K.; Ho, W.S.; et al. Rare Copy Number Variants in Tourette Syndrome Disrupt Genes in Histaminergic Pathways and Overlap with Autism. Biol. Psychiatry 2012, 71, 392–402. [Google Scholar] [CrossRef]
- Lei, J.; Deng, X.; Zhang, J.; Su, L.; Xu, H.; Liang, H.; Huang, X.; Song, Z.; Deng, H. Mutation screening of the HDC gene in Chinese Han patients with Tourette syndrome. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2011, 159B, 72–76. [Google Scholar] [CrossRef]
- Dong, H.; Liu, W.; Liu, M.; Xu, L.; Li, Q.; Zhang, R.; Zhang, X.; Liu, S. Investigation of a Possible Role for the Histidine Decarboxylase Gene in Tourette Syndrome in the Chinese Han Population: A Family-Based Study. PLoS ONE 2016, 11, e0160265. [Google Scholar] [CrossRef] [PubMed]
- Willsey, A.J.; Fernandez, T.V.; Yu, D.; King, R.A.; Dietrich, A.; Xing, J.; Sanders, S.J.; Mandell, J.D.; Huang, A.Y.; Richer, P.; et al. De Novo Coding Variants Are Strongly Associated with Tourette Disorder. Neuron 2017, 94, 486–499.e9. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mandell, J.D.; Kumar, Y.; Sun, N.; Morris, M.T.; Arbelaez, J.; Nasello, C.; Dong, S.; Duhn, C.; Zhao, X.; et al. De Novo Sequence and Copy Number Variants Are Strongly Associated with Tourette Disorder and Implicate Cell Polarity in Pathogenesis. Cell Rep. 2018, 24, 3441–3454.e12. [Google Scholar] [CrossRef]
- gnomAD. Genome Aggregation Database. Available online: https://gnomad.broadinstitute.org/ (accessed on 19 February 2021).
- Zhao, X.; Wang, S.; Hao, J.; Zhu, P.; Zhang, X.; Wu, M. A Whole-Exome Sequencing Study of Tourette Disorder in a Chinese Population. DNA Cell Biol. 2020, 39, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Tian, M.; He, F.; Li, J.; Xie, H.; Liu, W.; Zhang, Y.; Zhang, R.; Yi, M.; Che, F.; et al. Mutations in ASH1L confer susceptibility to Tourette syndrome. Mol. Psychiatry 2020, 25, 476–490. [Google Scholar] [CrossRef]
- Tsetsos, F.; Yu, D.; Sul, J.H.; Huang, A.Y.; Illmann, C.; Osiecki, L.; Darrow, S.M.; Hirschtritt, M.E.; Greenberg, E.; Muller-Vahl, K.R.; et al. Synaptic processes and immune-related pathways implicated in Tourette syndrome. Transl. Psychiatry 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Malhotra, D.; Sebat, J. CNVs: Harbingers of a Rare Variant Revolution in Psychiatric Genetics. Cell 2012, 148, 1223–1241. [Google Scholar] [CrossRef]
- Huang, A.Y.; Yu, D.; Davis, L.K.; Sul, J.H.; Tsetsos, F.; Ramensky, V.; Zelaya, I.; Ramos, E.M.; Osiecki, L.; Chen, J.A.; et al. Rare Copy Number Variants in NRXN1 and CNTN6 Increase Risk for Tourette Syndrome. Neuron 2017, 94, 1101–1111.e7. [Google Scholar] [CrossRef]
- Mercati, O.; Huguet, G.; Danckaert, A.; André-Leroux, G.; Maruani, A.; Bellinzoni, M.; Rolland, T.; Gouder, L.; Mathieu, A.; Buratti, J.; et al. CNTN6 mutations are risk factors for abnormal auditory sensory perception in autism spectrum disorders. Mol. Psychiatry 2017, 22, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Kashevarova, A.A.; Nazarenko, L.P.; Schultz-Pedersen, S.; Skryabin, N.A.; Salyukova, O.A.; Chechetkina, N.N.; Tolmacheva, E.N.; Rudko, A.A.; Magini, P.; Graziano, C.; et al. Single gene microdeletions and microduplication of 3p26.3 in three unrelated families: CNTN6 as a new candidate gene for intellectual disability. Mol. Cytogenet. 2014, 7, 97. [Google Scholar] [CrossRef] [PubMed]
- Pak, C.; Danko, T.; Zhang, Y.; Aoto, J.; Anderson, G.; Maxeiner, S.; Yi, F.; Wernig, M.; Südhof, T.C. Human Neuropsychiatric Disease Modeling using Conditional Deletion Reveals Synaptic Transmission Defects Caused by Heterozygous Mutations in NRXN1. Cell Stem Cell 2015, 17, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, S.K.; Huq, A.M.; Wilson, B.J.; Chugani, H.T. Tourette syndrome is associated with recurrent exonic copy number variants. Neurology 2010, 74, 1583–1590. [Google Scholar] [CrossRef]
- Nag, A.; Bochukova, E.; Kremeyer, B.; Campbell, D.; Muller, H.; Valencia-Duarte, A.V.; Cardona, J.; Rivas, I.C.; Mesa, S.C.; Cuartas, M.; et al. CNV Analysis in Tourette Syndrome Implicates Large Genomic Rearrangements in COL8A1 and NRXN1. PLoS ONE 2013, 8, e59061. [Google Scholar] [CrossRef]
- Glessner, J.T.; Wang, K.; Cai, G.; Korvatska, O.; Kim, C.E.; Wood, S.; Zhang, H.; Estes, A.; Brune, C.W.; Bradfield, J.P.; et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009, 459, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Ching, M.; Shen, Y.; Tan, W.-H.; Jeste, S.; Morrow, E.; Chen, X.; Mukaddes, N.M.; Yoo, S.-Y.; Hanson, E.; Hundley, R.; et al. Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2010, 153B, 937–947. [Google Scholar] [CrossRef]
- Kirov, G.; Gumus, D.; Chen, W.; Norton, N.; Georgieva, L.; Sari, M.; O’Donovan, M.C.; Erdogan, F.; Owen, M.J.; Ropers, H.-H.; et al. Comparative genome hybridization suggests a role for NRXN1 and APBA2 in schizophrenia. Hum. Mol. Genet. 2007, 17, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Eriguchi, Y.; Kuwabara, H.; Inai, A.; Kawakubo, Y.; Nishimura, F.; Kakiuchi, C.; Tochigi, M.; Ohashi, J.; Aoki, N.; Kato, K.; et al. Identification of candidate genes involved in the etiology of sporadic Tourette syndrome by exome sequencing. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2017, 174, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Nasello, C.; Deng, L.; Wang, N.; Zhang, Y.; Xu, Z.; Song, Z.; Kwan, K.; King, R.A.; Pang, Z.P.; et al. The PNKD gene is associated with Tourette Disorder or Tic disorder in a multiplex family. Mol. Psychiatry 2017, 23, 1487–1495. [Google Scholar] [CrossRef]
- Robertson, M.M. The Prevalence and Epidemiology of Gilles de La Tourette Syndrome. Part 1: The Epidemiological and Prevalence Studies. J. Psychosom. Res. 2008, 65, 461–472. [Google Scholar] [CrossRef]
- Carlson, C.S.; Matise, T.C.; North, K.E.; Haiman, C.A.; Fesinmeyer, M.; Buyske, S.; Schumacher, F.; Peters, U.; Franceschini, N.; Ritchie, M.D.; et al. Generalization and Dilution of Association Results from European GWAS in Populations of Non-European Ancestry: The PAGE Study. PLoS Biol. 2013, 11, e1001661. [Google Scholar] [CrossRef]
- Cavanna, A.E.; Rickards, H. The psychopathological spectrum of Gilles de la Tourette syndrome. Neurosci. Biobehav. Rev. 2013, 37, 1008–1015. [Google Scholar] [CrossRef]
- Anttila, V.; Bulik-Sullivan, B.; Finucane, H.K.; Walters, R.K.; Bras, J.; Duncan, L.; Escott-Price, V.; Falcone, G.J.; Gormley, P.; Malik, R.; et al. Analysis of shared heritability in common disorders of the brain. Science 2018, 360, eaap8757. [Google Scholar] [CrossRef]
- Lee, P.H.; Anttila, V.; Won, H.; Feng, Y.-C.A.; Rosenthal, J.; Zhu, Z.; Tucker-Drob, E.M.; Nivard, M.; Grotzinger, A.D.; Posthuma, D.; et al. Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders. Cell 2019, 179, 1469–1482.e11. [Google Scholar] [CrossRef] [PubMed]
- Mathews, C.; Darrow, S.; Hirschtritt, M.; Yu, N.; Davis, L.; Scharf, J. Symmetry and Disinhibition are Heritable Endophenotypes For Tourette Syndrome. Eur. Neuropsychopharmacol. 2019, 29, S738. [Google Scholar] [CrossRef]
- Yang, Z.; Wu, H.; Lee, P.H.; Tsetsos, F.; Davis, L.K.; Yu, D.; Lee, S.H.; Dalsgaard, S.; Haavik, J.; Barta, C.; et al. Investigating Shared Genetic Basis Across Tourette Syndrome and Comorbid Neurodevelopmental Disorders Along the Impulsivity-Compulsivity Spectrum. Biol. Psychiatry 2021, 90, 317–327. [Google Scholar] [CrossRef]
- Eapen, V.; Robertson, M. Are there distinct subtypes in Tourette syndrome? Pure-Tourette syndrome versus Tourette syndrome-plus, and simple versus complex tics. Neuropsychiatr. Dis. Treat. 2015, 11, 1431–1436. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levy, A.M.; Paschou, P.; Tümer, Z. Candidate Genes and Pathways Associated with Gilles de la Tourette Syndrome—Where Are We? Genes 2021, 12, 1321. https://doi.org/10.3390/genes12091321
Levy AM, Paschou P, Tümer Z. Candidate Genes and Pathways Associated with Gilles de la Tourette Syndrome—Where Are We? Genes. 2021; 12(9):1321. https://doi.org/10.3390/genes12091321
Chicago/Turabian StyleLevy, Amanda M., Peristera Paschou, and Zeynep Tümer. 2021. "Candidate Genes and Pathways Associated with Gilles de la Tourette Syndrome—Where Are We?" Genes 12, no. 9: 1321. https://doi.org/10.3390/genes12091321
APA StyleLevy, A. M., Paschou, P., & Tümer, Z. (2021). Candidate Genes and Pathways Associated with Gilles de la Tourette Syndrome—Where Are We? Genes, 12(9), 1321. https://doi.org/10.3390/genes12091321