Pathogenic DNM1L Variant (1085G>A) Linked to Infantile Progressive Neurological Disorder: Evidence of Maternal Transmission by Germline Mosaicism and Influence of a Contemporary in cis Variant (1535T>C)

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
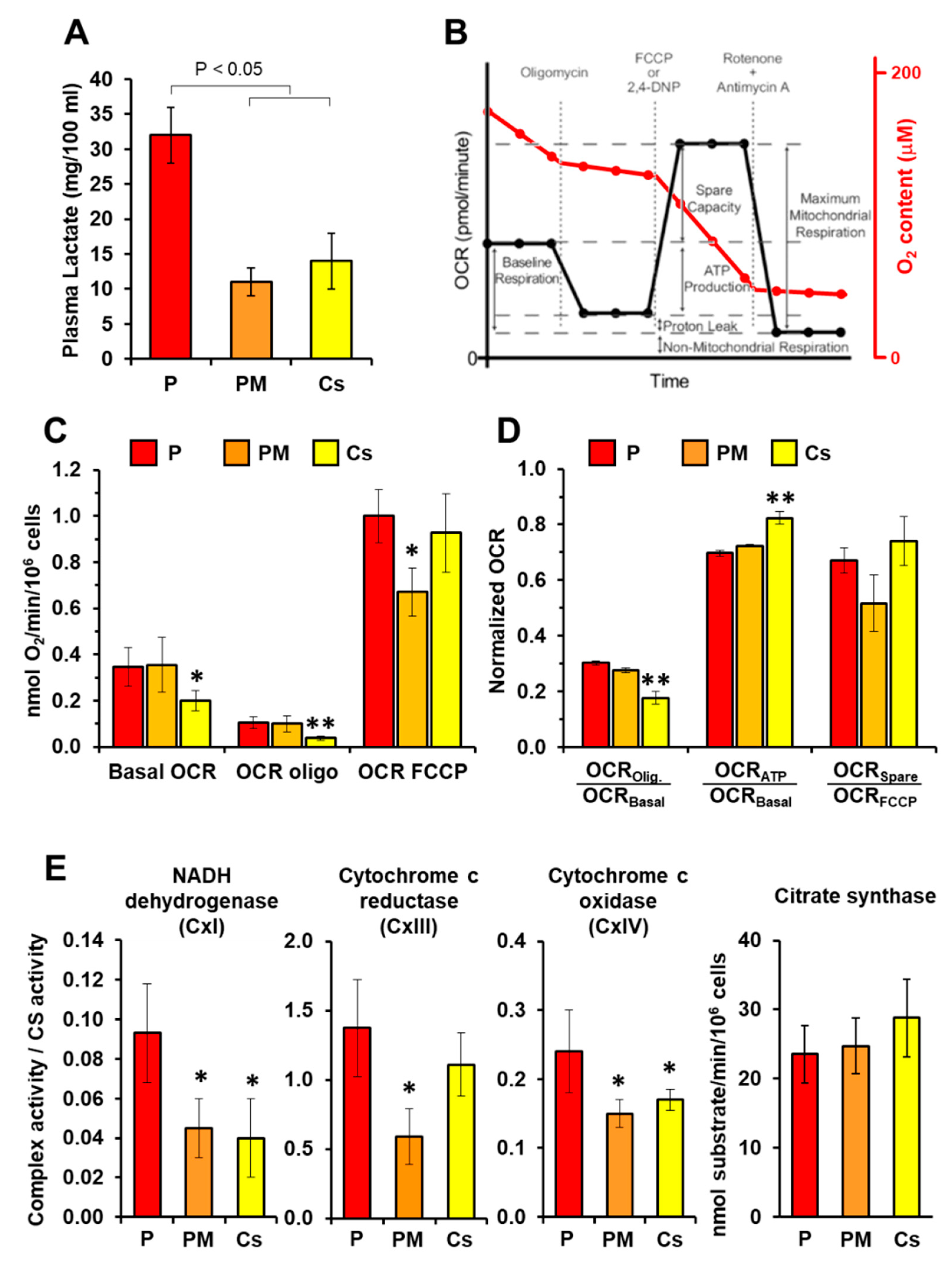
2.2. Measurement of Respiratory Activity
2.3. Measurement of Enzymatic Activities
2.4. Live Cell Imaging of ROS, Mitochondrial Membrane Potential, and Ca2+
2.5. Reverse Transcription-Polymerase Chain Reaction
2.6. Western Blotting Analysis
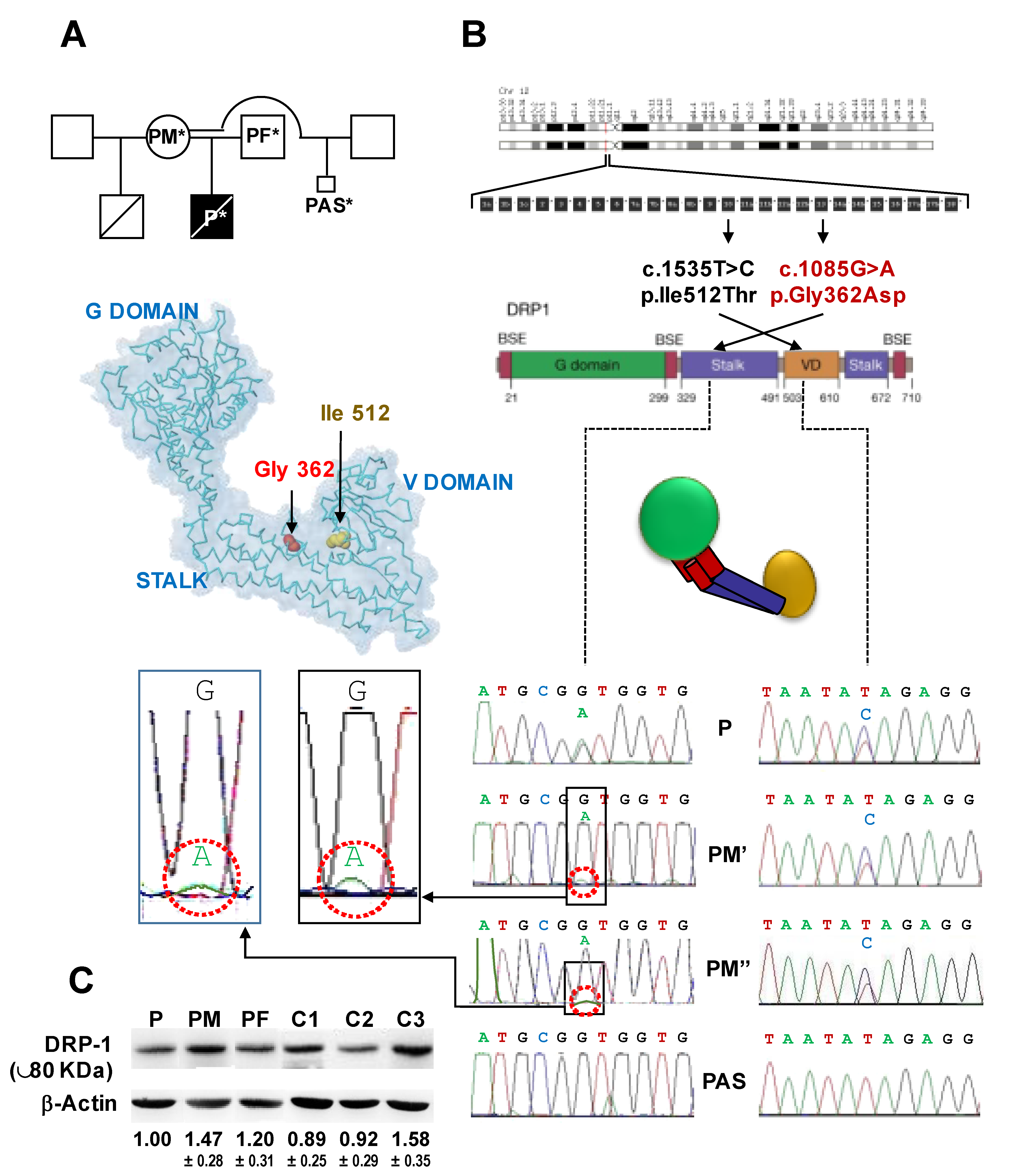
2.7. DNM1L Gene Sequencing
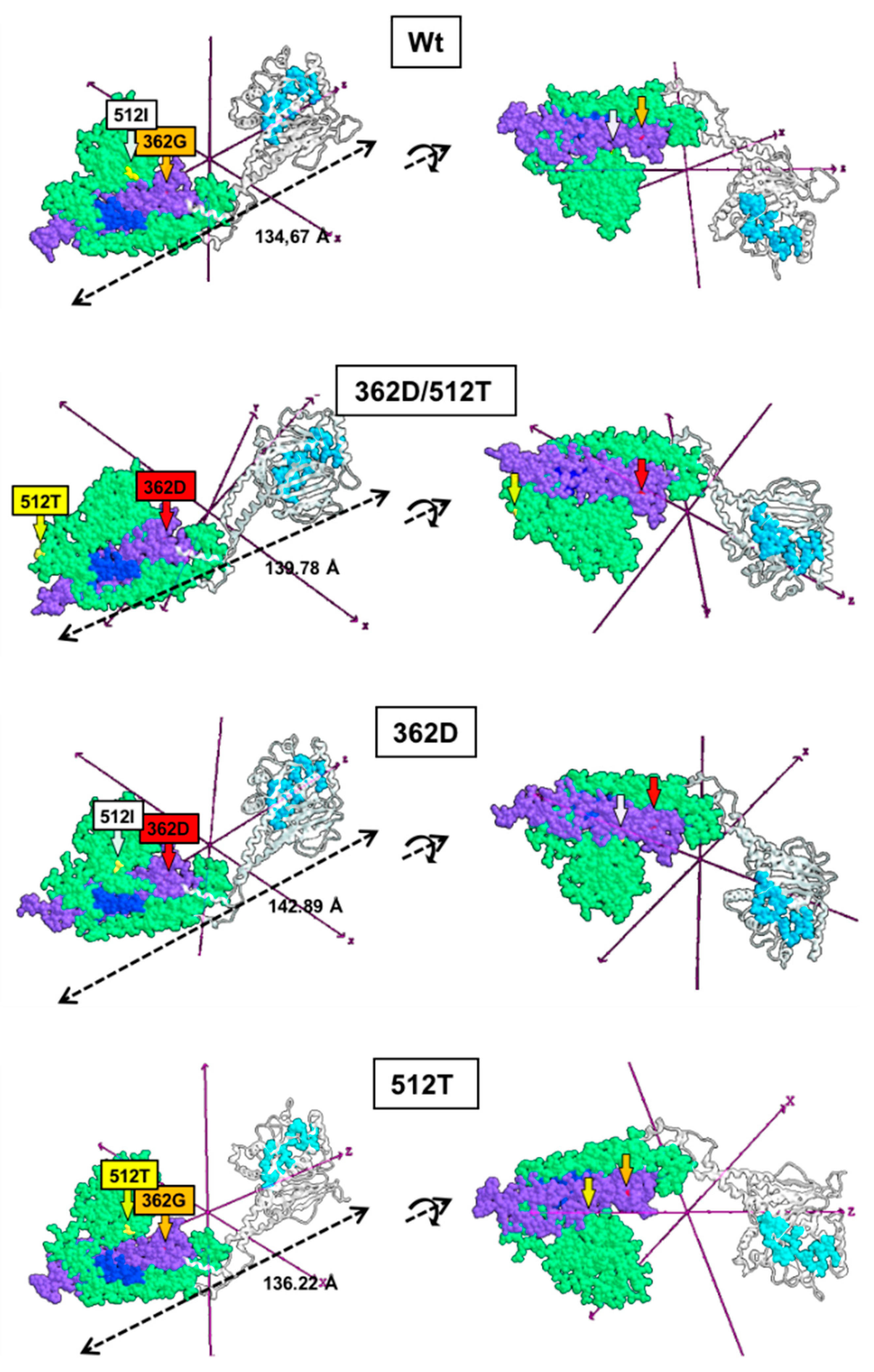
2.8. Structural Modelling of the Wt and Mutated Drp1
2.9. Statistical Analysis
3. Results
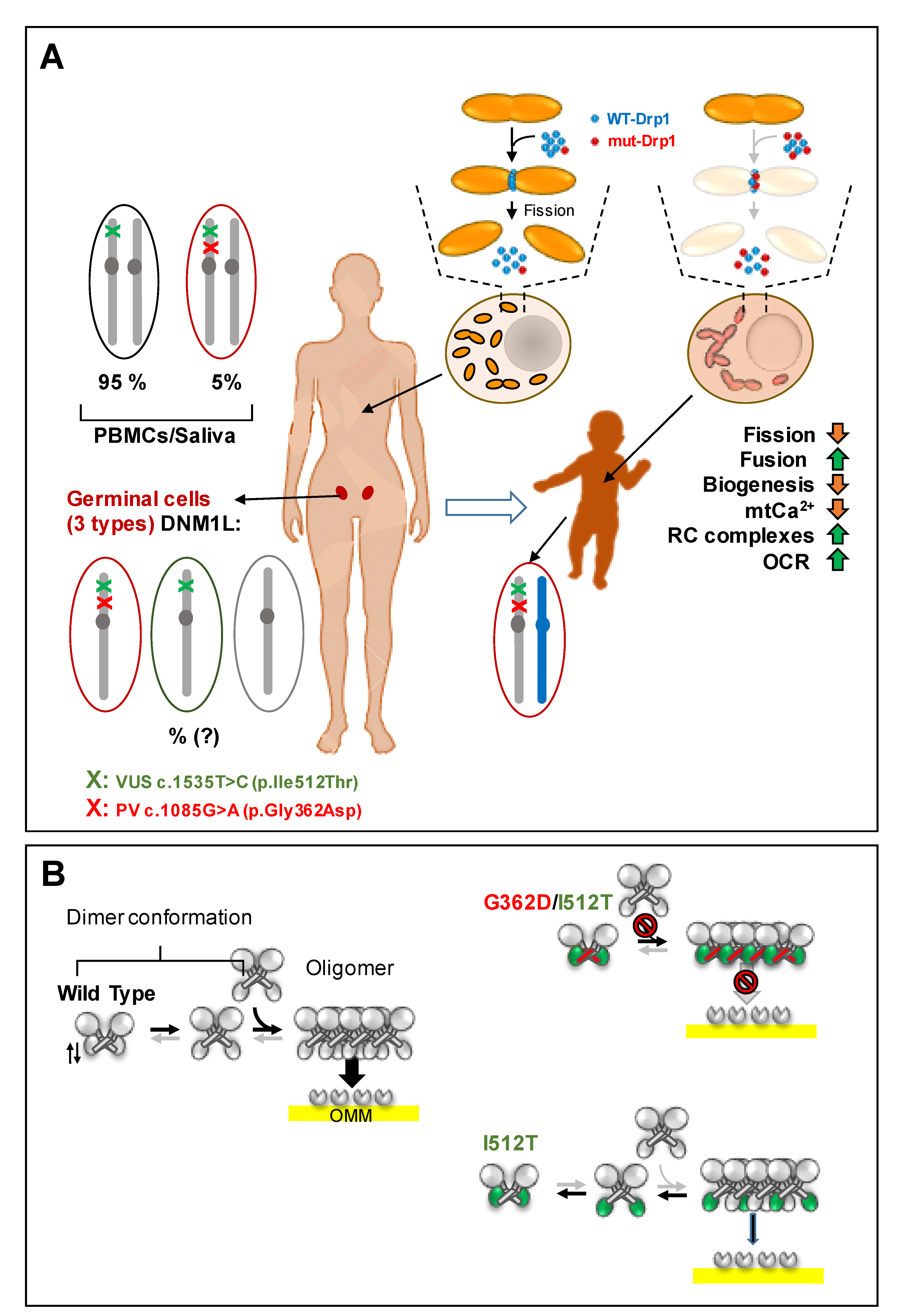
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef]
- Dorn, G.W., 2nd. Evolving Concepts of Mitochondrial Dynamics. Annu. Rev. Physiol. 2019, 81, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kraus, F.; Roy, K.; Pucadyil, T.J.; Ryan, M.T. Function and regulation of the divisome for mitochondrial fission. Nature 2021, 590, 57–66. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef]
- Bertholet, A.M.; Delerue, T.; Millet, A.M.; Moulis, M.F.; David, C.; Daloyau, M.; Arnauné-Pelloquin, L.; Davezac, N.; Mils, V.; Miquel, M.C.; et al. Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol Dis. 2016, 90, 3–19. [Google Scholar] [CrossRef]
- Di Nottia, M.; Verrigni, D.; Torraco, A.; Rizza, T.; Bertini, E.; Carrozzo, R. Mitochondrial Dynamics: Molecular Mechanisms, Related Primary Mitochondrial Disorders and Therapeutic Approaches. Genes 2021, 12, 247. [Google Scholar] [CrossRef] [PubMed]
- Waterham, H.R.; Koster, J.; van Roermund, C.W.; Mooyer, P.A.; Wanders, R.J.; Leonard, J.V. A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 2007, 356, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- Vanstone, J.R.; Smith, A.M.; McBride, S.; Naas, T.; Holcik, M.; Antoun, G.; Harper, M.E.; Michaud, J.; Sell, E.; Chakraborty, P.; et al. DNM1L-related mitochondrial fission defect presenting as refractory epilepsy. Eur. J. Hum. Genet. 2016, 24, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Sheffer, R.; Douiev, L.; Edvardson, S.; Shaag, A.; Tamimi, K.; Soiferman, D.; Meiner, V.; Saada, A. Postnatal microcephaly and pain insensitivity due to a de novo heterozygous DNM1L mutation causing impaired mitochondrial fission and function. Am. J. Med. Genet. A 2016, 170, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Yoon, G.; Malam, Z.; Paton, T.; Marshall, C.R.; Hyatt, E.; Ivakine, Z.; Scherer, S.W.; Lee, K.S.; Hawkins, C.; Cohn, R.D.; et al. Lethal Disorder of Mitochondrial Fission Caused by Mutations in DNM1L. J. Pediatr. 2016, 171, 313–316.e2. [Google Scholar] [CrossRef]
- Nasca, A.; Legati, A.; Baruffini, E.; Nolli, C.; Moroni, I.; Ardissone, A.; Goffrini, P.; Ghezzi, D. Biallelic Mutations in DNM1L are Associated with a Slowly Progressive Infantile Encephalopathy. Hum. Mutat. 2016, 37, 898–903. [Google Scholar] [CrossRef]
- Verrigni, D.; Di Nottia, M.; Ardissone, A.; Baruffini, E.; Nasca, A.; Legati, A.; Bellacchio, E.; Fagiolari, G.; Martinelli, D.; Fusco, L.; et al. Clinical-genetic features and peculiar muscle histopathology in infantile DNM1L-related mitochondrial epileptic encephalopathy. Hum. Mutat. 2019, 40, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Resta, N.; Susca, F.C.; Di Giacomo, M.C.; Stella, A.; Bukvic, N.; Bagnulo, R.; Simone, C.; Guanti, G. A homozygous frameshift mutation in the ESCO2 gene: Evidence of intertissue and interindividual variation in Nmd efficiency. J. Cell Physiol. 2006, 209, 67–73. [Google Scholar] [CrossRef]
- Barrientos, A.; Fontanesi, F.; Díaz, F. Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays. Curr. Protoc. Hum. Genet. 2009. [Google Scholar] [CrossRef] [PubMed]
- Trunzo, R.; Santacroce, R.; D’Andrea, G.; Longo, V.; De Girolamo, G.; Dimatteo, C.; Leccese, A.; Lillo, V.; Papadia, F.; Margaglione, M. Mutation analysis in hyperphenylalaninemia patients from South Italy. Clin. Biochem. 2013, 46, 1896–1898. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Finkel, T.; Menazza, S.; Holmström, K.M.; Parks, R.J.; Liu, J.; Sun, J.; Liu, J.; Pan, X.; Murphy, E. The ins and outs of mitochondrial calcium. Circ. Res. 2015, 116, 1810–1819. [Google Scholar] [CrossRef]
- Lu, B.; Kennedy, B.; Clinton, R.W.; Wang, E.J.; McHugh, D.; Stepanyants, N.; Macdonald, P.J.; Mears, J.A.; Qi, X.; Ramachandran, R. Steric interference from intrinsically disordered regions controls dynamin-related protein 1 self-assembly during mitochondrial fission. Sci. Rep. 2018, 8, 10879. [Google Scholar] [CrossRef]
- Chou, C.H.; Lin, C.C.; Yang, M.C.; Wei, C.C.; Liao, H.D.; Lin, R.C.; Tu, W.Y.; Kao, T.C.; Hsu, C.M.; Cheng, J.T.; et al. GSK3beta-mediated Drp1 phosphorylation induced elongated mitochondrial morphology against oxidative stress. PLoS ONE 2012, 7, e49112. [Google Scholar]
- Fröhlich, C.; Grabiger, S.; Schwefel, D.; Faelber, K.; Rosenbaum, E.; Mears, J.; Rocks, O.; Daumke, O. Structural insights into oligomerization and mitochondrial remodelling of dynamin 1-like protein. EMBO J. 2013, 32, 1280–1292. [Google Scholar] [CrossRef]
- Lightowlers, R.N.; Taylor, R.W.; Turnbull, D.M. Mutations causing mitochondrial disease: What is new and what challenges remain? Science 2015, 349, 1494–1499. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef]
- Ishihara, N.; Nomura, M.; Jofuku, A.; Kato, H.; Suzuki, S.O.; Masuda, K.; Otera, H.; Nakanishi, Y.; Nonaka, I.; Goto, Y.; et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 2009, 11, 958–966. [Google Scholar] [CrossRef]
- Wakabayashi, J.; Zhang, Z.; Wakabayashi, N.; Tamura, Y.; Fukaya, M.; Kensler, T.W.; Iijima, M.; Sesaki, H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 2009, 186, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.M.; Yuan, B.; Robberecht, C.; Pfundt, R.; Szafranski, P.; McEntagart, M.E.; Nagamani, S.C.; Erez, A.; Bartnik, M.; Wiśniowiecka-Kowalnik, B.; et al. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am. J. Hum. Genet. 2014, 95, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Donkervoort, S.; Hu, Y.; Stojkovic, T.; Voermans, N.C.; Foley, A.R.; Leach, M.E.; Dastgir, J.; Bolduc, V.; Cullup, T.; de Becdelièvre, A.; et al. Mosaicism for dominant collagen 6 mutations as a cause for intrafamilial phenotypic variability. Hum. Mutat. 2015, 36, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Berko, E.R.; Suzuki, M.; Beren, F.; Lemetre, C.; Alaimo, C.M.; Calder, R.B.; Ballaban-Gil, K.; Gounder, B.; Kampf, K.; Kirschen, J.; et al. Mosaic epigenetic dysregulation of ectodermal cells in autism spectrum disorder. PLoS Genet. 2014, 10, e1004402. [Google Scholar] [CrossRef] [PubMed]
- Laurentino, S.; Beygo, J.; Nordhoff, V.; Kliesch, S.; Wistuba, J.; Borgmann, J.; Buiting, K.; Horsthemke, B.; Gromoll, J. Epigenetic germline mosaicism in infertile men. Hum. Mol. Genet. 2015, 24, 1295–1304. [Google Scholar] [CrossRef]
- Benard, G.; Rossignol, R. Ultrastructure of the mitochondrion and its bearing on function and bioenergetics. Antioxid. Redox Signal. 2008, 10, 1313–1342. [Google Scholar] [CrossRef]
- Baker, N.; Patel, J.; Khacho, M. Linking mitochondrial dynamics, cristae remodeling and supercomplex formation: How mitochondrial structure can regulate bioenergetics. Mitochondrion 2019, 49, 259–268. [Google Scholar] [CrossRef]
- Burman, J.L.; Pickles, S.; Wang, C.; Sekine, S.; Vargas, J.N.S.; Zhang, Z.; Youle, A.M.; Nezich, C.L.; Wu, X.; Hammer, J.A.; et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 2017, 216, 3231–3247. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Chakrabarti, O. Mitochondrial hyperfusion: A friend or a foe. Biochem. Soc. Trans. 2020, 48, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Simoni, A.M.; Bianchi, K.; De Stefani, D.; Leo, S.; Wieckowski, M.R.; Rizzuto, R. Mitochondrial dynamics and Ca2+ signaling. Biochim. Biophys. Acta 2006, 1763, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Csordás, G.; Weaver, D.; Hajnóczky, G. Endoplasmic Reticulum-Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Tugolukova, E.A.; Campbell, R.A.; Hoerger, K.B.; Schwerz, H.; Weyrich, A.S.; Rowley, J.W. Mitochondrial fission protein Drp1 regulates megakaryocyte and platelet mitochondrial morphology, platelet numbers, and platelet function. Blood 2017, 130 (Suppl. S1), 455. [Google Scholar]
- Lewis, S.C.; Uchiyama, L.F.; Nunnari, J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 2016, 353, aaf5549. [Google Scholar] [CrossRef]
- Chapman, J.; Ng, Y.S.; Nicholls, T.J. The Maintenance of Mitochondrial DNA Integrity and Dynamics by Mitochondrial Membranes. Life 2020, 10, 164. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Falkenberg, M.; Rantanen, A.; Park, C.B.; Gaspari, M.; Hultenby, K.; Rustin, P.; Gustafsson, C.M.; Larsson, N.G. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet. 2004, 13, 935–944. [Google Scholar] [CrossRef]
- Reubold, T.F.; Faelber, K.; Plattner, N.; Posor, Y.; Ketel, K.; Curth, U.; Schlegel, J.; Anand, R.; Manstein, D.J.; Noé, F.; et al. Crystal structure of the dynamin tetramer. Nature 2015, 525, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Kalia, R.; Wang, R.Y.; Yusuf, A.; Thomas, P.V.; Agard, D.A.; Shaw, J.M.; Frost, A. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 2018, 558, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Clinton, R.W.; Francy, C.A.; Ramachandran, R.; Qi, X.; Mears, J.A. Dynamin-related Protein 1 Oligomerization in Solution Impairs Functional Interactions with Membrane-anchored Mitochondrial Fission Factor. J. Biol. Chem. 2016, 291, 478–492. [Google Scholar] [CrossRef]
- Adaniya, S.M.; O-Uchi, J.; Cypress, M.W.; Kusakari, Y.; Jhun, B.S. Posttranslational modifications of mitochondrial fission and fusion proteins in cardiac physiology and pathophysiology. Am. J. Physiol. Cell Physiol. 2019, 316, C583–C604. [Google Scholar] [CrossRef] [PubMed]
- Rahit, K.M.T.H.; Tarailo-Graovac, M. Genetic Modifiers and Rare Mendelian Disease. Genes 2020, 11, 239. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piccoli, C.; Scrima, R.; D’Aprile, A.; Chetta, M.; Cela, O.; Pacelli, C.; Ripoli, M.; D’Andrea, G.; Margaglione, M.; Bukvic, N.; et al. Pathogenic DNM1L Variant (1085G>A) Linked to Infantile Progressive Neurological Disorder: Evidence of Maternal Transmission by Germline Mosaicism and Influence of a Contemporary in cis Variant (1535T>C). Genes 2021, 12, 1295. https://doi.org/10.3390/genes12091295
Piccoli C, Scrima R, D’Aprile A, Chetta M, Cela O, Pacelli C, Ripoli M, D’Andrea G, Margaglione M, Bukvic N, et al. Pathogenic DNM1L Variant (1085G>A) Linked to Infantile Progressive Neurological Disorder: Evidence of Maternal Transmission by Germline Mosaicism and Influence of a Contemporary in cis Variant (1535T>C). Genes. 2021; 12(9):1295. https://doi.org/10.3390/genes12091295
Chicago/Turabian StylePiccoli, Claudia, Rosella Scrima, Annamaria D’Aprile, Massimiliano Chetta, Olga Cela, Consiglia Pacelli, Maria Ripoli, Giovanna D’Andrea, Maurizio Margaglione, Nenad Bukvic, and et al. 2021. "Pathogenic DNM1L Variant (1085G>A) Linked to Infantile Progressive Neurological Disorder: Evidence of Maternal Transmission by Germline Mosaicism and Influence of a Contemporary in cis Variant (1535T>C)" Genes 12, no. 9: 1295. https://doi.org/10.3390/genes12091295
APA StylePiccoli, C., Scrima, R., D’Aprile, A., Chetta, M., Cela, O., Pacelli, C., Ripoli, M., D’Andrea, G., Margaglione, M., Bukvic, N., & Capitanio, N. (2021). Pathogenic DNM1L Variant (1085G>A) Linked to Infantile Progressive Neurological Disorder: Evidence of Maternal Transmission by Germline Mosaicism and Influence of a Contemporary in cis Variant (1535T>C). Genes, 12(9), 1295. https://doi.org/10.3390/genes12091295