
Development and Evaluation of the Ancestry Informative Marker Panel of the VISAGE Basic Tool

 , , , ,
, , , ,  , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection and Balancing of Component Ancestry Informative SNPs
2.2. Compilation of a 4132-Sample Population Dataset
2.3. Genotyping Concordance amongst MPS Sequence Data and Online Databases
2.4. Evaluation of Ancestry and Co-Ancestry Inference Efficiency of the BT AIMs
2.5. Evaluation of Ancestry and Co-Ancestry Inference Efficiency of the BT AIMs
3. Results and Discussion
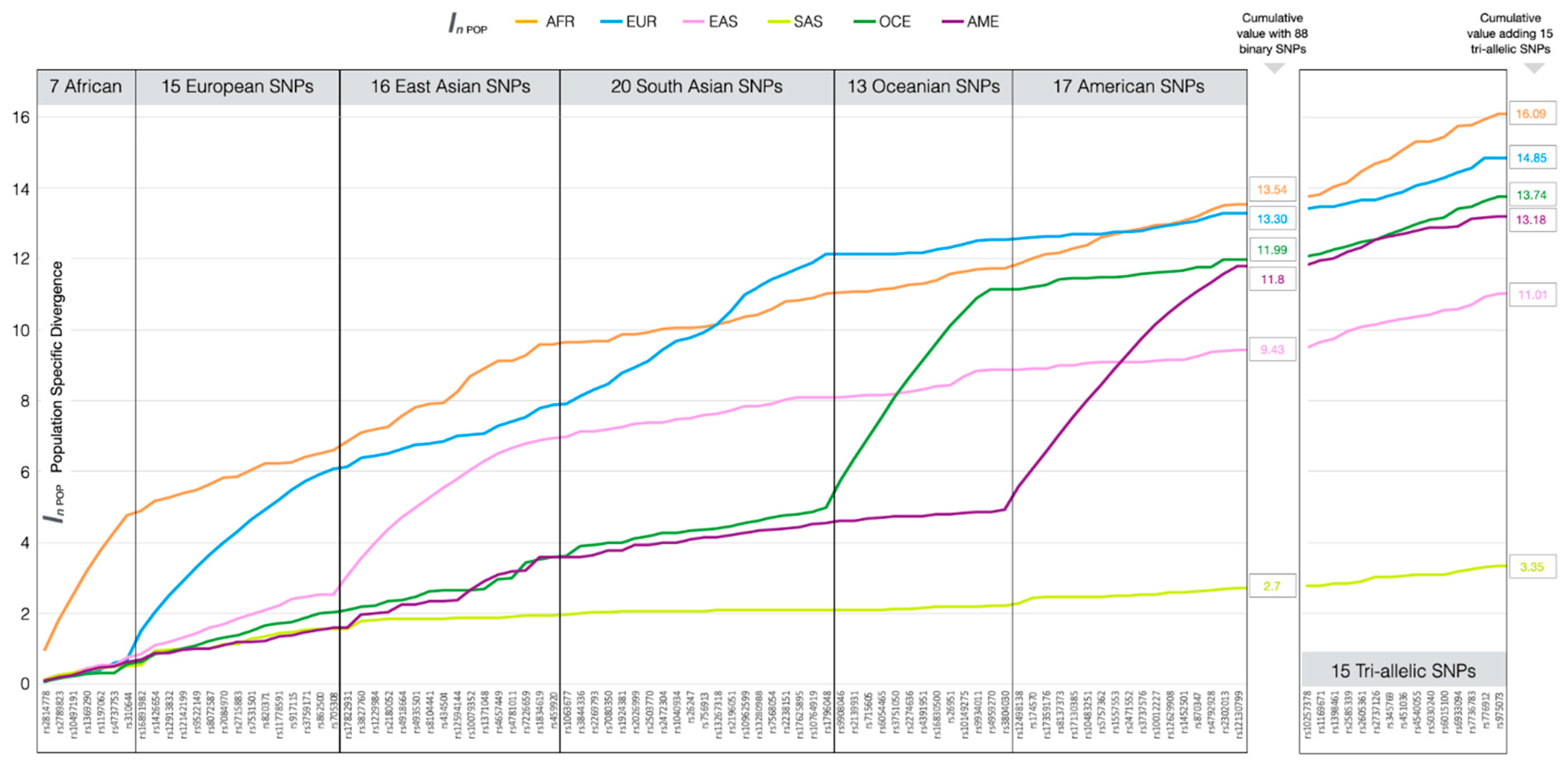
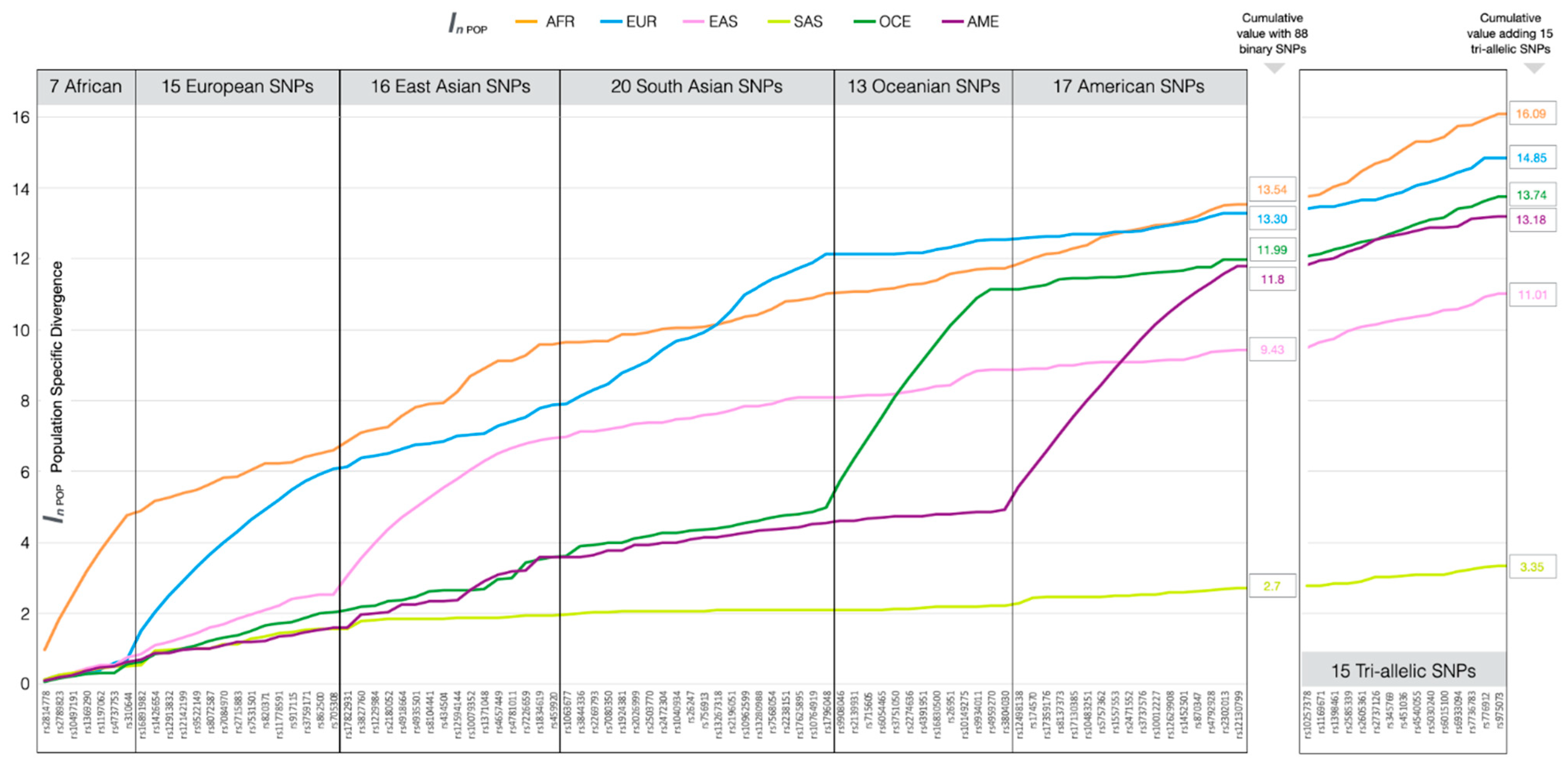
3.1. Ancestry Informative SNPs in BT
3.2. Compilation of Reference and Test Population Datasets
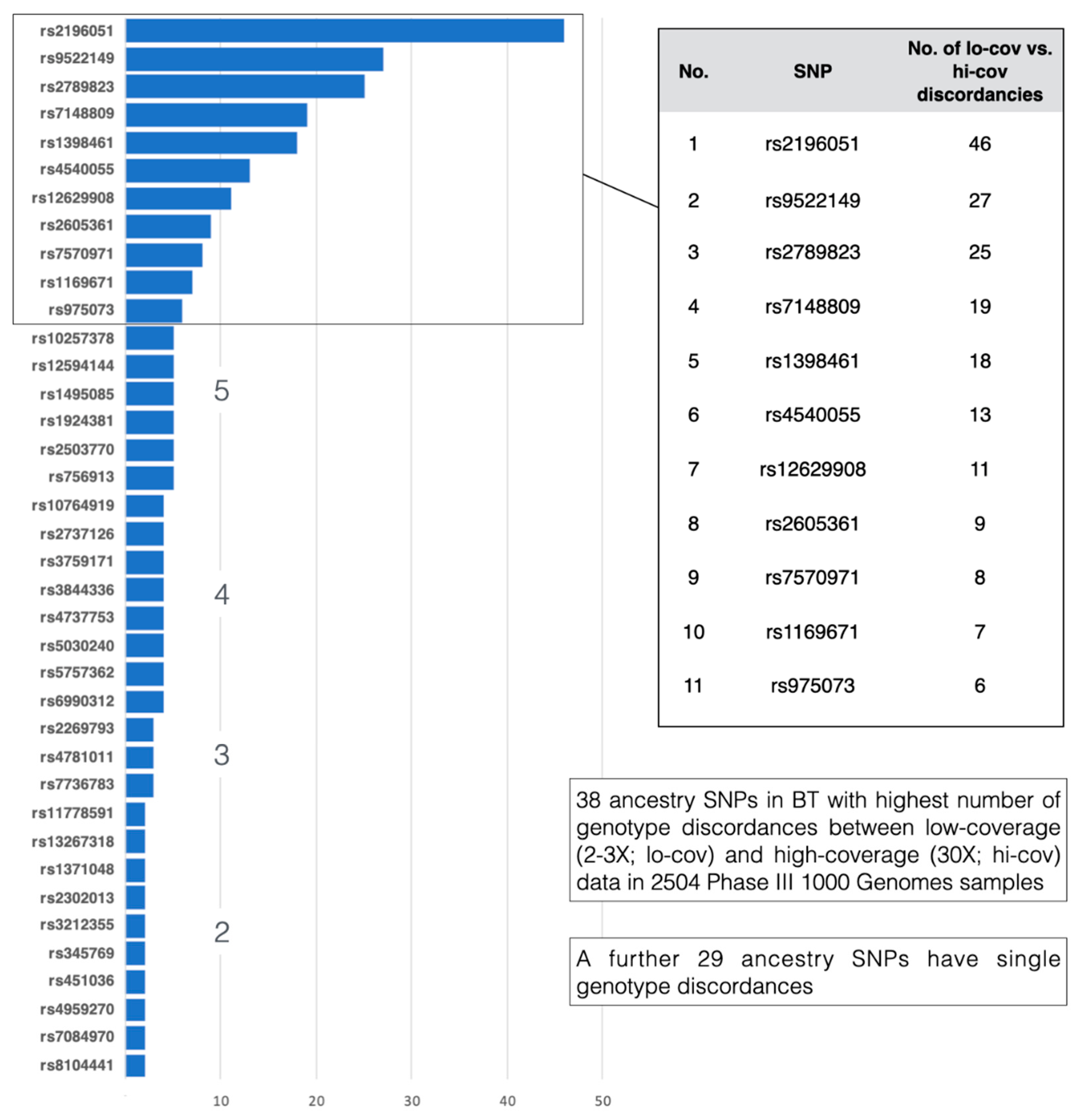
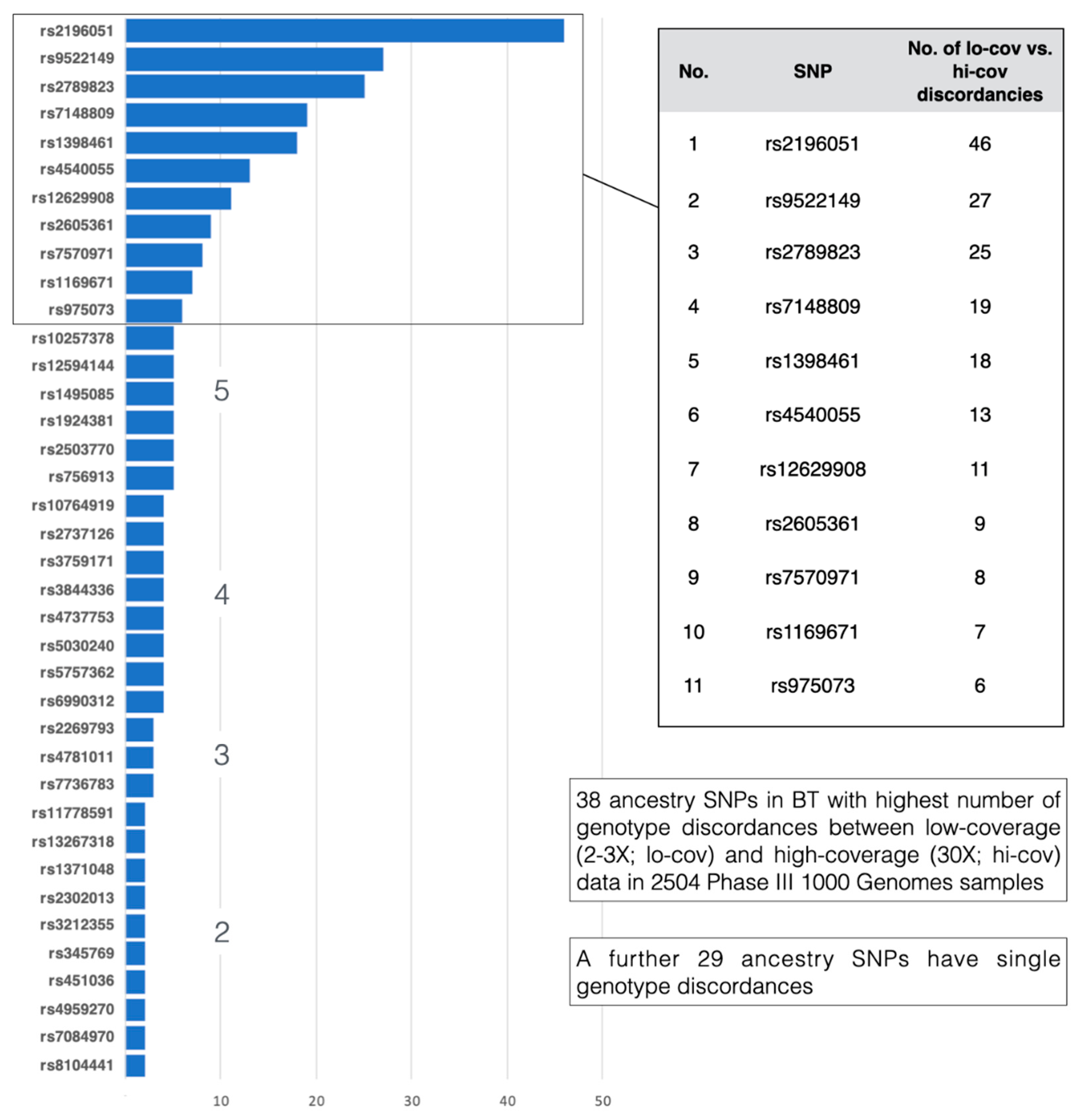
3.3. Genotyping Concordance
3.3.1. HGDP-CEPH in-House BT Genotypes vs. 1000 Genomes Whole-Genome-Sequence Data
3.3.2. 1000 Genomes Phase 3 Genotypes vs. 1000 Genomes High Sequence Coverage Genotypes
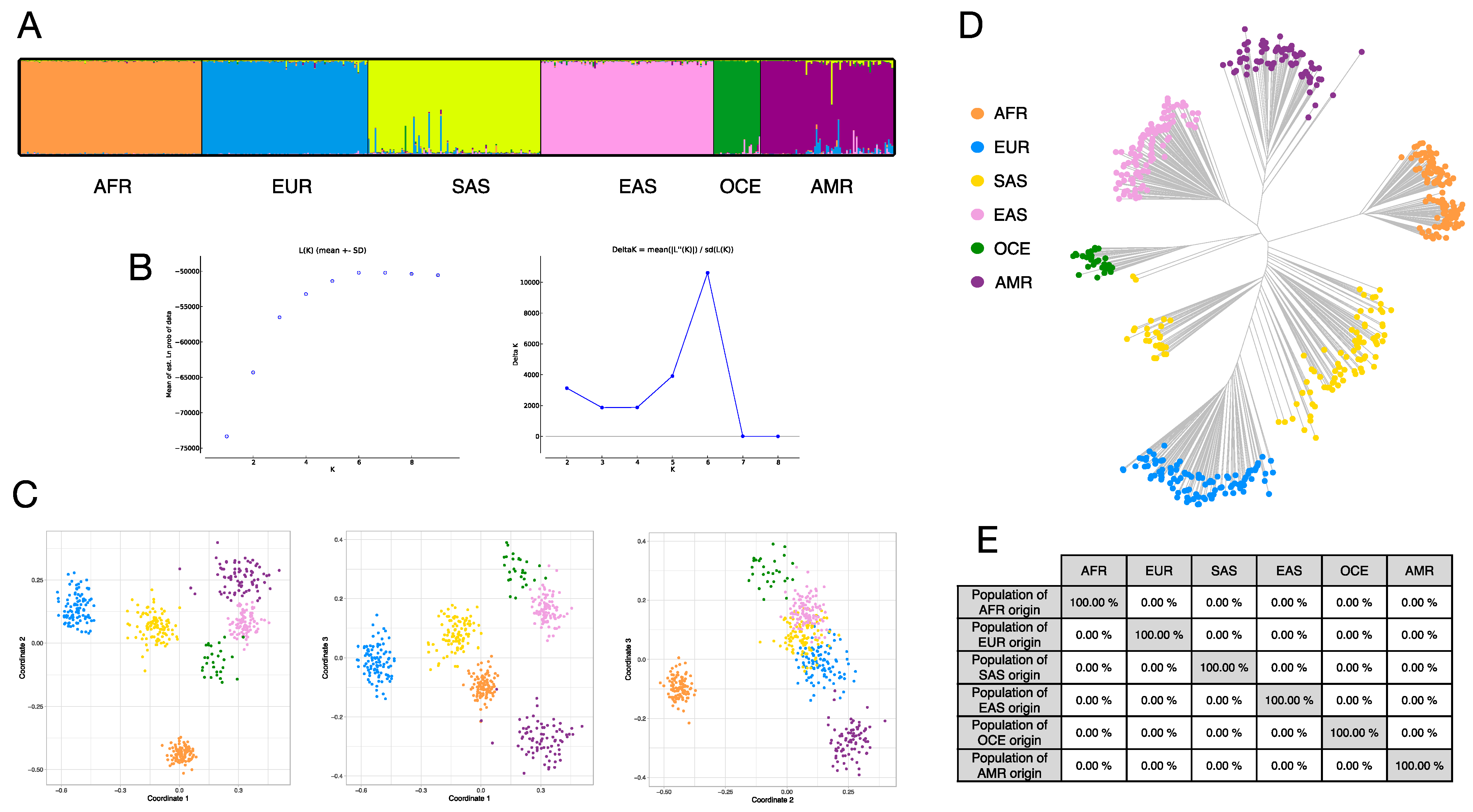
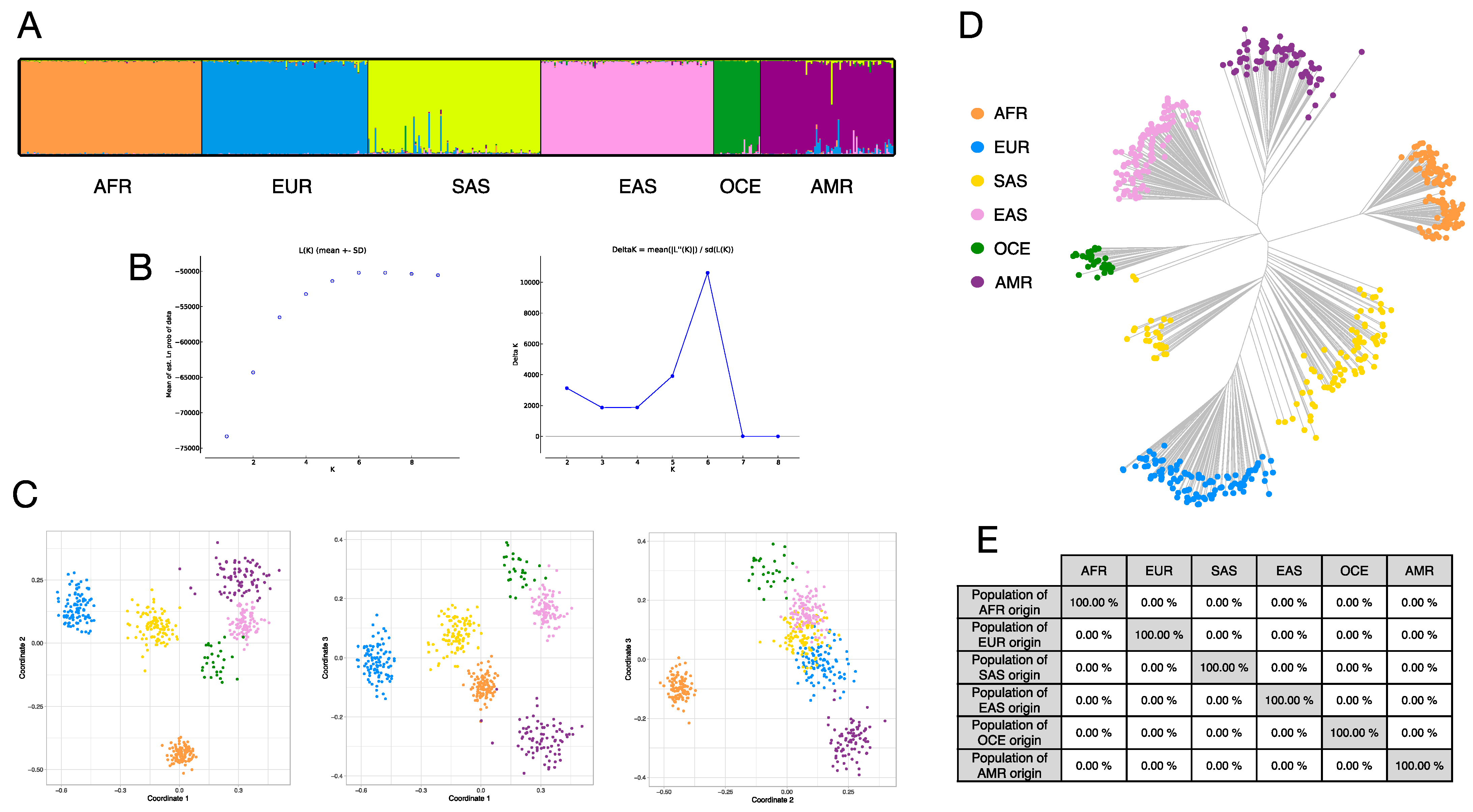
3.4. Ancestry Inference Efficiency of the BT Ancestry SNPs
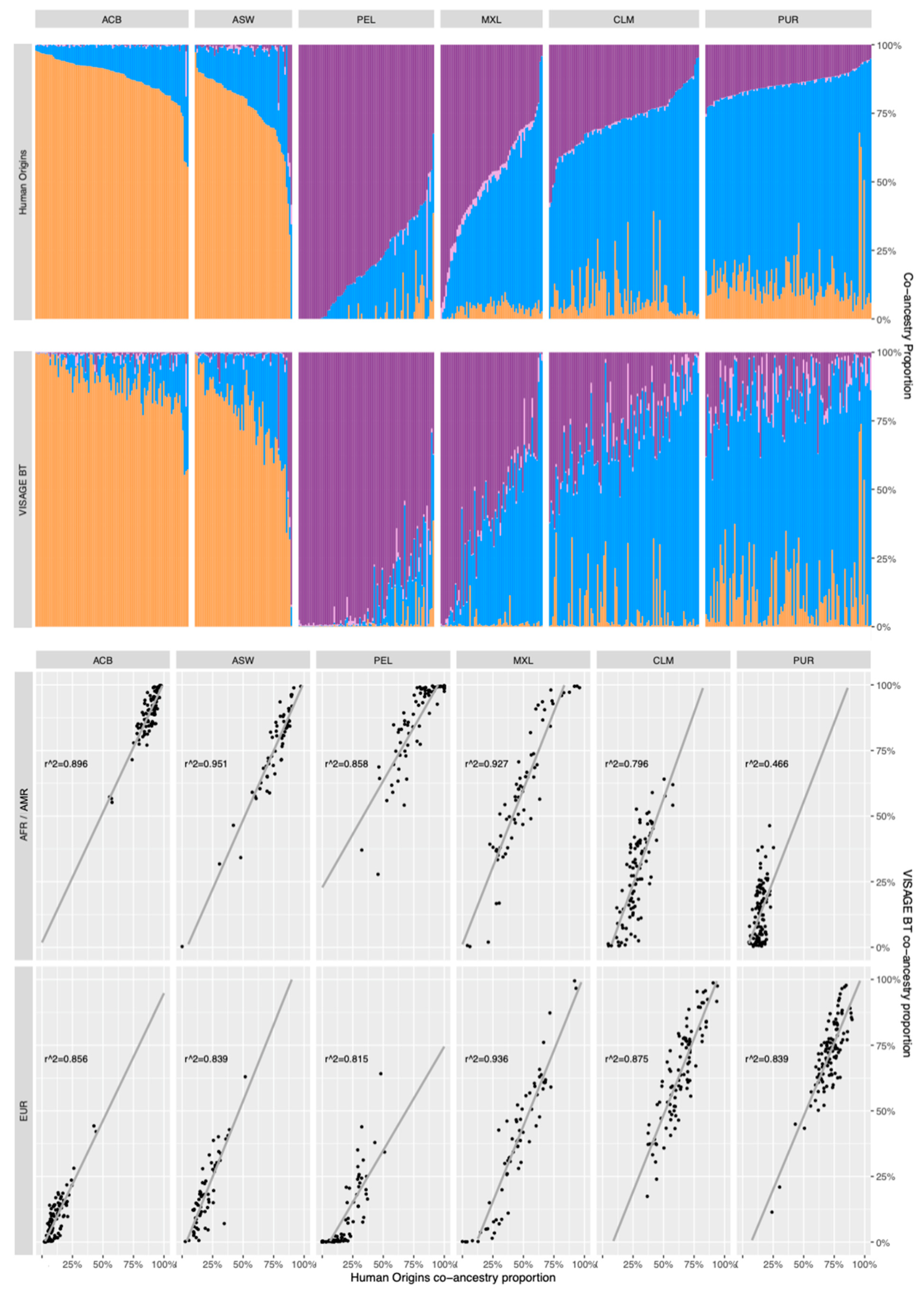
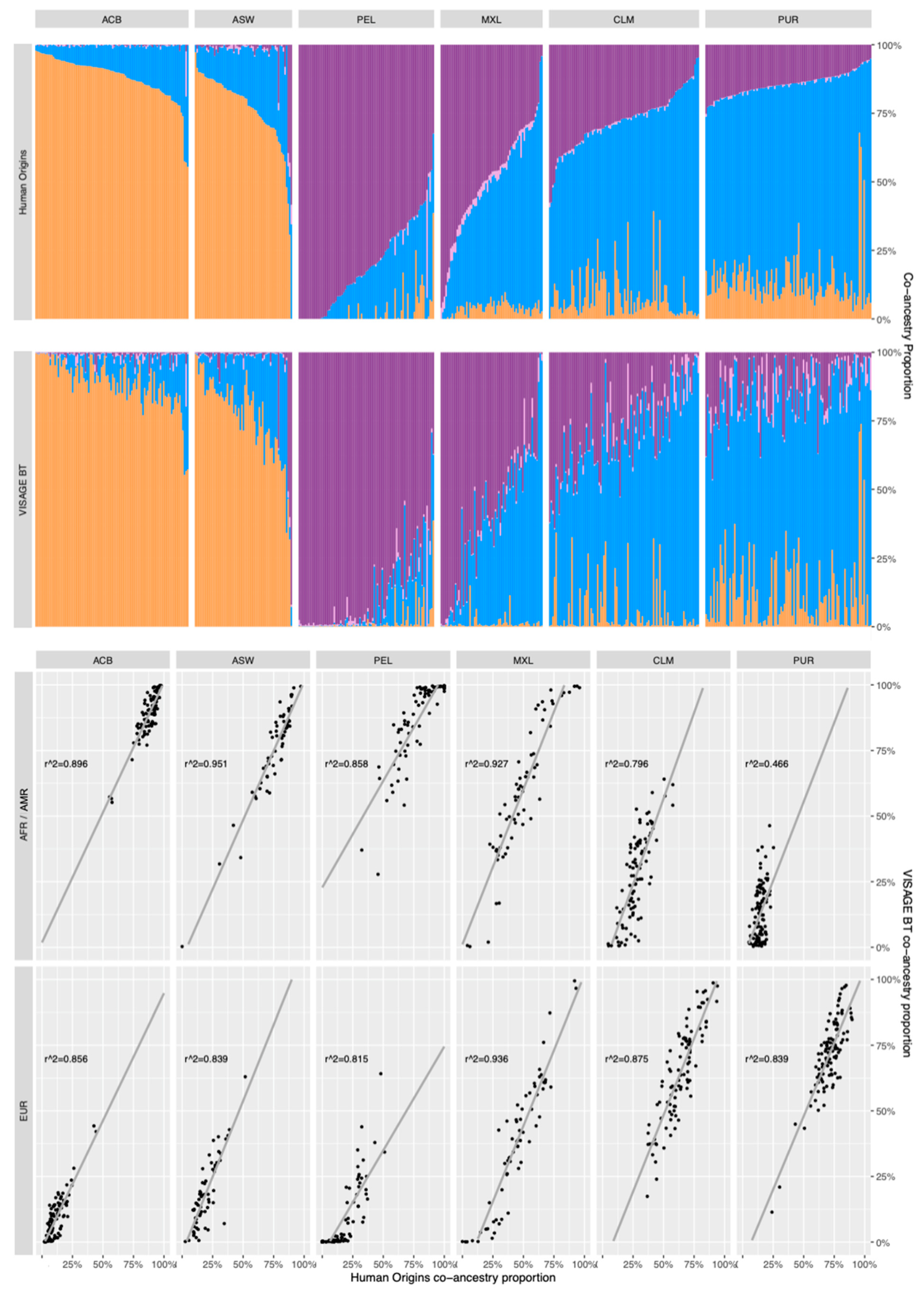
3.5. Co-Ancestry Analysis of Known Admixed Individuals in 1000 Genomes with BT Ancestry SNPs
3.5.1. Comparison of Co-Ancestry Patterns Obtained with the Human Origins Panel and BT
3.5.2. Using GDA as a Simple Snipper-Based Evaluation of Admixture
3.5.3. Comparisons of 10-Percentile Co-Ancestry Ratio Patterns
3.6. Audit of Additional Variation in BT SNPs from the GnomAD 3.1 Database
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
- Erasmus University Medical Center Rotterdam, Rotterdam, The Netherlands: Manfred Kayser, Vivian Kalamara, Arwin Ralf, Athina Vidaki
- Jagiellonian University, Krakow, Poland: Wojciech Branicki, Ewelina Pośpiech, Aleksandra Pisarek
- Universidade de Santiago de Compostela, Santiago de Compostela, Spain: Ángel Carracedo, Maria Victoria Lareu, Christopher Phillips, Ana Freire-Aradas, Ana Mosquera-Miguel, María de la Puente
- Medizinische Universität Innsbruck, Innsbruck, Austria: Walther Parson, Catarina Xavier, Antonia Heidegger, Harald Niederstätter
- Universität zu Köln, Cologne, Germany: Michael Nothnagel, Maria-Alexandra Katsara, Tarek Khellaf
- King’s College London, London, UK: Barbara Prainsack, Gabrielle Samuel
- Klinikum der Universität zu Köln, Cologne, Germany: Peter M. Schneider, Theresa E. Gross,
- Bundeskriminalamt, Wiesbaden, Germany: Ingo Bastisch, Nathalie Schury, Jens Teodoridis,
- Institut National de Police Scientifique, Lyon, France: François-Xavier Laurent, Caroline
- Netherlands Forensic Institute, The Hague, Netherlands: Titia Sijen, Kris van der Gaag,
- National Forensic Centre, Swedish Police Authority, Linköping, Sweden: Johannes Hedman,
- Metropolitan Police Service, London, United Kingdom: Shazia Khan, Carole E. Ames, Andrew Revoir
- Centralne Laboratorium Kryminalistyczne Policji, Warsaw, Poland: Magdalena Spólnicka, Ewa Kartasinska, Anna Woźniak
References
- Phillips, C. Forensic genetic analysis of bio-geographical ancestry. Forensic Sci. Int. Genet. 2015, 18, 49–65. [Google Scholar] [CrossRef]
- Kayser, M. Forensic DNA Phenotyping: Predicting human appearance from crime scene material for investigative purposes. Forensic Sci. Int. Genet. 2015, 18, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Freire-Aradas, A.; Phillips, C.; Lareu, M.V. Forensic individual age estimation with DNA: From initial approaches to methylation tests. Forensic Sci. Rev. 2017, 29, 121–144. [Google Scholar] [PubMed]
- King, T.E.; Fortes, G.G.; Balaresque, P.; Thomas, M.G.; Balding, D.; Delser, P.M.; Neumann, R.; Parson, W.; Knapp, M.; Walsh, S.; et al. Identification of the remains of King Richard III. Nat. Commun. 2014, 5, 5631. [Google Scholar] [CrossRef]
- Walsh, S.; Liu, F.; Ballantyne, K.N.; van Oven, M.; Lao, O.; Kayser, M. IrisPlex: A sensitive DNA tool for accurate prediction of blue and brown eye colour in the absence of ancestry information. Forensic Sci. Int. Genet. 2011, 5, 170–180. [Google Scholar] [CrossRef]
- Walsh, S.; Liu, F.; Wollstein, A.; Kovatsi, L.; Ralf, A.; Kosiniak-Kamysz, A.; Branicki, W.; Kayser, M. The HIrisPlex system for simultaneous prediction of hair and eye colour from DNA. Forensic Sci. Int. Genet. 2013, 7, 98–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, S.; Chaitanya, L.; Breslin, K.; Muralidharan, C.; Bronikowska, A.; Pospiech, E.; Koller, J.; Kovatsi, L.; Wollstein, A.; Branicki, W.; et al. Global skin colour prediction from DNA. Hum. Genet. 2017, 136, 847–863. [Google Scholar] [CrossRef] [Green Version]
- Phillips, C.; Parson, W.; Lundsberg, B.; Santos, C.; Freire-Aradas, A.; Torres, M.; Eduardoff, M.; Børsting, C.; Johansen, P.; Fondevila, M.; et al. Building a forensic ancestry panel from the ground up: The EUROFORGEN Global AIM-SNP set. Forensic Sci. Int. Genet. 2014, 11, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Phillips, C.; Freire Aradas, A.; Kriegel, A.K.; Fondevila, M.; Bulbul, O.; Santos, C.; Serrulla Rech, F.; Perez Carceles, M.D.; Carracedo, A.; Schneider, P.M.; et al. Eurasiaplex: A forensic SNP assay for differentiating European and South Asian ancestries. Forensic Sci. Int. Genet. 2013, 7, 359–366. [Google Scholar] [CrossRef]
- Carvalho Gontijo, C.; Porras-Hurtado, L.G.; Freire-Aradas, A.; Fondevila, M.; Santos, C.; Salas, A.; Henao, J.; Isaza, C.; Beltrán, L.; Nogueira Silbiger, V.; et al. PIMA: A population informative multiplex for the Americas. Forensic Sci. Int. Genet. 2020, 44, 102200. [Google Scholar] [CrossRef] [Green Version]
- Santos, C.; Phillips, C.; Fondevila, M.; Daniel, R.; van Oorschot, R.A.H.; Burchard, E.G.; Schanfield, M.S.; Souto, L.J.; Uacyisrael, J.; Via, M.; et al. Pacifiplex: An ancestry-informative SNP panel centred on Australia and the Pacific region. Forensic Sci. Int. Genet. 2016, 20, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.; McNevin, D.; Kidd, K.K.; Lagacé, R.; Wootton, S.; de la Puente, M.; Freire-Aradas, A.; Mosquera-Miguel, A.; Eduardoff, M.; Gross, T.; et al. MAPlex-A massively parallel sequencing ancestry analysis multiplex for Asia-Pacific populations. Forensic Sci. Int. Genet. 2019, 42, 213–226. [Google Scholar] [CrossRef] [Green Version]
- Breslin, K.; Wills, B.; Ralf, A.; Ventayol Garcia, M.; Kukla-Bartoszek, M.; Pospiech, E.; Freire-Aradas, A.; Xavier, C.; Ingold, S.; de La Puente, M.; et al. HIrisPlex-S system for eye, hair, and skin color prediction from DNA: Massively parallel sequencing solutions for two common forensically used platforms. Forensic Sci. Int. Genet. 2019, 43, 102152. [Google Scholar] [CrossRef] [PubMed]
- Al-Asfi, M.; McNevin, D.; Mehta, B.; Power, D.; Gahan, M.E.; Daniel, R. Assessment of the Precision ID Ancestry Panel. Int. J. Legal. Med. 2018, 132, 1581–1594. [Google Scholar] [CrossRef]
- The 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- Bergström, A.; McCarthy, S.A.; Hui, R.; Almarri, M.A.; Ayub, Q.; Danecek, P.; Chen, Y.; Felkel, S.; Hallast, P.; Kamm, J.; et al. Insights into human genetic variation and population history from 929 diverse genomes. Science 2020, 367, 1339–1349. [Google Scholar] [CrossRef]
- Byrska-Bishop, M.; Evani, U.S.; Zhao, X.; Basile, A.O.; Abel, H.J.; Regier, A.A.; André Corvelo, A.; Clarke, W.E.; Musunuri, R.; Nagulapalli, K.; et al. High Coverage Whole Genome Sequencing of the Expanded 1000 Genomes Project Cohort Including 602 trios. Available online: https://www.internationalgenome.org/data-portal/data-collection/30x-grch38; http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/working/20190425_NYGC_GATK/ (accessed on 20 June 2021).
- Xavier, C.; de la Puente, M.; Phillips, C.; Eduardoff, M.; Heidegger, A.; Mosquera-Miguel, A.; Freire-Aradas, A.; Lagace, R.; Wootton, S.; Power, D.; et al. Forensic evaluation of the Asia Pacific ancestry-informative MAPlex assay. Forensic Sci. Int. Genet. 2020, 48, 102344. [Google Scholar] [CrossRef]
- Cheung, E.Y.Y.; Phillips, C.; Eduardoff, M.; Lareu, M.V.; McNevin, D. Performance of ancestry-informative SNP and microhaplotype markers. Forensic Sci. Int. Genet. 2019, 43, 102141. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.; Amigo, J.; Tillmar, A.O.; Peck, M.A.; de la Puente, M.; Ruiz-Ramírez, J.; Bittner, F.; Idrizbegović, Š.; Wang, Y.; Parsons, T.J.; et al. A compilation of tri-allelic SNPs from 1000 Genomes and use of the most polymorphic loci for a large-scale human identification panel. Forensic Sci. Int. Genet. 2020, 46, 102232. [Google Scholar] [CrossRef] [Green Version]
- Galanter, J.M.; Fernandez-Lopez, J.C.; Gignoux, C.R.; Barnholtz-Sloan, J.; Fernandez-Rozadilla, C.; Via, M.; Hidalgo-Miranda, A.; Contreras, A.V.; Figueroa, L.U.; Raska, P.; et al. Development of a panel of genome-wide ancestry informative markers to study admixture throughout the Americas. PLoS Genet. 2012, 8, e1002554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidd, K.K.; Speed, W.C.; Pakstis, A.J.; Furtado, M.R.; Fang, R.; Madbouly, A.; Maiers, M.; Middha, M.; Friedlaender, F.R.; Kidd, J.R. Progress toward an efficient panel of SNPs for ancestry inference. Forensic Sci. Int. Genet. 2014, 10, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Pereira, V.; Freire-Aradas, A.; Ballard, D.; Børsting, C.; Diez, V.; Pruszkowska-Przybylska, P.; Ribeiro, J.; Achakzai, N.M.; Aliferi, A.; Bulbul, O.; et al. Development and validation of the EUROFORGEN NAME (North African and Middle Eastern) ancestry panel. Forensic Sci. Int. Genet. 2019, 42, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Bigham, A.W.; Mei, R.; Gutierrez, G.; Weiss, K.M.; Brutsaert, T.D.; Leon-Velarde, F.; Moore, L.G.; Vargas, E.; McKeigue, P.M.; et al. A genomewide admixture mapping panel for Hispanic/Latino populations. Am. J. Hum. Genet 2007, 80, 1171–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: http://mathgene.usc.es/Snipper/ (accessed on 20 June 2021).
- Phillips, C.; Amigo, J.; McNevin, D.; de la Puente, M.; Cheung, E.Y.Y.; Lareu, M.V. Online population data resources for forensic SNP analysis with Massively Parallel Sequencing: An overview of online population data for forensic purposes. In Forensic DNA Analysis: Technological Development and Innovative Applications; Pilli, E., Berti, A., Eds.; CRC Press: Boca Raton, FL, USA, 2021. [Google Scholar]
- Mallick, S.; Li, H.; Lipson, M.; Mathieson, I.; Gymrek, M.; Racimo, F.; Zhao, M.; Chennagiri, N.; Nordenfelt, S.; Tandon, A.; et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 2016, 538, 201–206. [Google Scholar] [CrossRef]
- Pagani, L.; Lawson, D.J.; Jagoda, E.; Mörseburg, A.; Eriksson, A.; Mitt, M.; Clemente, F.; Hudjashov, G.; DeGiorgio, M.; Saag, L.; et al. Genomic analyses inform on migration events during the peopling of Eurasia. Nature 2016, 538, 238–242. [Google Scholar] [CrossRef] [Green Version]
- Xavier, C.; de la Puente, M.; Mosquera-Miguel, A.; Freire-Aradas, A.; Kalamara, V.; Vidaki, A.; Gross, T.; Revoir, A.; Pośpiech, E.; Kartasińska, E.; et al. Development and validation of the VISAGE AmpliSeq Basic Tool to predict appearance and ancestry from DNA. Forensic Sci. Int. Genet. 2020, 48, 102336. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R: A Language and Environment for Statistical Computing. Available online: http://www.r-project.org/ (accessed on 20 June 2021).
- Paradis, E. Pegas: An R package for population genetics with an integrated-modular approach. Bioinformatics 2010, 26, 419–420. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, E.Y.Y.; Gahan, M.E.; McNevin, D. Prediction of biogeographical ancestry from genotype: A comparison of classifiers. Int. J. Legal Med. 2017, 131, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Palencia-Madrid, L.; Xavier, C.; de la Puente, M.; Hohoff, C.; Phillips, C.; Kayser, M.; Parson, W. on behalf of the VISAGE Consortium, Evaluation of the VISAGE Basic Tool for Appearance and Ancestry Prediction Using PowerSeq Chemistry on the MiSeq FGx System. Genes 2020, 11, 708. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Pop. | SNP | KK/PIAP | gAIMs | LACE | Other | No. | Pop. | SNP | KK/PIAP | gAIMs | LACE | Other | No. | Pop. | SNP | KK/PIAP | gAIMs | LACE | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | AFR | rs10497191 | Kiddlab | - | LACE | 1 | AME | rs10012227 | - | gAIMs | - | 1 | Eurasia | rs1495085 | - | - | - | NAME | ||
| 2 | AFR | rs1197062 | - | gAIMs | LACE | 2 | AME | rs10483251 | - | gAIMs | LACE | 2 | Eurasia | rs1757928 | - | - | - | NAME | ||
| 3 | AFR | rs1369290 | - | gAIMs | - | 3 | AME | rs12130799 | PIAP | - | - | 3 | Eurasia | rs2337024 | - | - | - | NAME | ||
| 4 | AFR | rs2789823 | - | gAIMs | - | 4 | AME | rs12498138 | Kiddlab | gAIMs | - | 4 | Eurasia | rs6989963 | - | - | - | NAME | ||
| 5 | AFR | rs2814778 | Kiddlab | gAIMs | - | 5 | AME | rs12629908 | PIAP | - | - | 5 | Eurasia | rs6990312 | Kiddlab | - | - | |||
| 6 | AFR | rs310644 | Kiddlab | gAIMs | - | 6 | AME | rs1452501 | - | gAIMs | LACE | 6 | Eurasia | rs7148809 | - | - | - | NAME | ||
| 7 | AFR | rs4737753 | - | - | - | NAME | 7 | AME | rs1557553 | - | gAIMs | LACE | 7 | Eurasia | rs12203115 | - | - | - | NAME | |
| 1 | EUR | rs11778591 | - | gAIMs | LACE | 8 | AME | rs17130385 | - | gAIMs | LACE | 8 | Eurasia | rs2227203 | - | - | - | NAME | ||
| 2 | EUR | rs12142199 | - | gAIMs | - | 9 | AME | rs17359176 | - | gAIMs | LACE | 9 | Eurasia | rs39897 | - | - | - | Eurasiaplex | ||
| 3 | EUR | rs12913832 | Kiddlab | gAIMs | - | 10 | AME | rs174570 | Kiddlab | gAIMs | LACE | 10 | Eurasia | rs4308478 | - | - | - | NAME | ||
| 4 | EUR | rs1426654 | Kiddlab | gAIMs | - | 11 | AME | rs2302013 | - | gAIMs | - | 11 | Eurasia | rs7570971 | - | - | - | NAME | ||
| 5 | EUR | rs16891982 | Kiddlab | gAIMs | - | 12 | AME | rs2471552 | - | gAIMs | - | 12 | Eurasia | rs984038 | - | - | - | NAME | ||
| 6 | EUR | rs2715883 | - | gAIMs | - | 13 | AME | rs3737576 | Kiddlab | - | - | 1 | SAS | rs1040934 | - | - | - | Shriver | ||
| 7 | EUR | rs3759171 | - | gAIMs | LACE | 14 | AME | rs4792928 | - | gAIMs | - | 2 | SAS | rs1063677 | - | - | - | Shriver | ||
| 8 | EUR | rs705308 | PIAP | - | - | 15 | AME | rs5757362 | - | - | LACE | 3 | SAS | rs10764919 | - | - | LACE | |||
| 9 | EUR | rs7084970 | - | gAIMs | - | 16 | AME | rs8137373 | - | gAIMs | - | 4 | SAS | rs10962599 | - | - | - | Eurasiaplex | ||
| 10 | EUR | rs7531501 | - | gAIMs | - | 17 | AME | rs870347 | Kiddlab | - | - | 5 | SAS | rs13267318 | - | - | - | Shriver | ||
| 11 | EUR | rs8072587 | - | gAIMs | - | 1 | EAS | rs10079352 | - | gAIMs | LACE | 6 | SAS | rs13280988 | - | - | LACE | |||
| 12 | EUR | rs820371 | - | gAIMs | LACE | 2 | EAS | rs1229984 | Kiddlab | gAIMs | - | 7 | SAS | rs17625895 | - | - | - | Eurasiaplex | ||
| 13 | EUR | rs862500 | - | gAIMs | LACE | 3 | EAS | rs12594144 | - | gAIMs | - | 8 | SAS | rs1796048 | - | - | - | Shriver | ||
| 14 | EUR | rs917115 | Kiddlab | gAIMs | - | 4 | EAS | rs1371048 | - | gAIMs | - | 9 | SAS | rs1924381 | - | gAIMs | LACE | |||
| 15 | EUR | rs9522149 | Kiddlab | gAIMs | - | 5 | EAS | rs17822931 | - | gAIMs | - | 10 | SAS | rs2026999 | - | - | - | Shriver | ||
| 1 | OCE | rs10149275 | - | gAIMs | - | 6 | EAS | rs1834619 | Kiddlab | gAIMs | - | 11 | SAS | rs2196051 | Kiddlab | - | - | Eurasiaplex | ||
| 2 | OCE | rs16830500 | - | gAIMs | - | 7 | EAS | rs2180052 | - | gAIMs | - | 12 | SAS | rs2238151 | Kiddlab | - | - | |||
| 3 | OCE | rs2139931 | - | gAIMs | - | 8 | EAS | rs3827760 | Kiddlab | gAIMs | - | 13 | SAS | rs2269793 | PIAP | - | - | |||
| 4 | OCE | rs2274636 | - | gAIMs | - | 9 | EAS | rs434504 | - | gAIMs | - | 14 | SAS | rs2472304 | - | - | - | Eurasiaplex | ||
| 5 | OCE | rs26951 | - | gAIMs | - | 10 | EAS | rs459920 | Kiddlab | - | - | 15 | SAS | rs2503770 | - | gAIMs | LACE | |||
| 6 | OCE | rs3751050 | - | gAIMs | - | 11 | EAS | rs4657449 | - | gAIMs | - | 16 | SAS | rs26247 | - | - | - | Shriver | ||
| 7 | OCE | rs3804030 | - | gAIMs | - | 12 | EAS | rs4781011 | PIAP | - | - | 17 | SAS | rs3844336 | - | - | - | Shriver | ||
| 8 | OCE | rs4391951 | - | gAIMs | - | 13 | EAS | rs4918664 | Kiddlab | gAIMs | - | 18 | SAS | rs7080350 | - | - | LACE | |||
| 9 | OCE | rs4959270 | - | x | - | Pacifiplex * | 14 | EAS | rs4935501 | - | gAIMs | - | 19 | SAS | rs7568054 | - | - | LACE | ||
| 10 | OCE | rs6054465 | - | gAIMs | - | 15 | EAS | rs7226659 | Kiddlab | - | - | 20 | SAS | rs756913 | - | - | - | Eurasiaplex | ||
| 11 | OCE | rs715605 | - | gAIMs | - | 16 | EAS | rs8104441 | - | gAIMs | - | |||||||||
| 12 | OCE | rs9908046 | - | gAIMs | - | MYH15 º | EAS | rs6437783 | - | gAIMs | - | - | ||||||||
| 13 | OCE | rs9934011 | - | gAIMs | - | OCA2 º | EAS | rs1800414 | Kiddlab | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de la Puente, M.; Ruiz-Ramírez, J.; Ambroa-Conde, A.; Xavier, C.; Pardo-Seco, J.; Álvarez-Dios, J.; Freire-Aradas, A.; Mosquera-Miguel, A.; Gross, T.E.; Cheung, E.Y.Y.; et al. Development and Evaluation of the Ancestry Informative Marker Panel of the VISAGE Basic Tool. Genes 2021, 12, 1284. https://doi.org/10.3390/genes12081284
de la Puente M, Ruiz-Ramírez J, Ambroa-Conde A, Xavier C, Pardo-Seco J, Álvarez-Dios J, Freire-Aradas A, Mosquera-Miguel A, Gross TE, Cheung EYY, et al. Development and Evaluation of the Ancestry Informative Marker Panel of the VISAGE Basic Tool. Genes. 2021; 12(8):1284. https://doi.org/10.3390/genes12081284
Chicago/Turabian Stylede la Puente, María, Jorge Ruiz-Ramírez, Adrián Ambroa-Conde, Catarina Xavier, Jacobo Pardo-Seco, Jose Álvarez-Dios, Ana Freire-Aradas, Ana Mosquera-Miguel, Theresa E. Gross, Elaine Y. Y. Cheung, and et al. 2021. "Development and Evaluation of the Ancestry Informative Marker Panel of the VISAGE Basic Tool" Genes 12, no. 8: 1284. https://doi.org/10.3390/genes12081284
APA Stylede la Puente, M., Ruiz-Ramírez, J., Ambroa-Conde, A., Xavier, C., Pardo-Seco, J., Álvarez-Dios, J., Freire-Aradas, A., Mosquera-Miguel, A., Gross, T. E., Cheung, E. Y. Y., Branicki, W., Nothnagel, M., Parson, W., Schneider, P. M., Kayser, M., Carracedo, Á., Lareu, M. V., Phillips, C., & on behalf of the VISAGE Consortium. (2021). Development and Evaluation of the Ancestry Informative Marker Panel of the VISAGE Basic Tool. Genes, 12(8), 1284. https://doi.org/10.3390/genes12081284